1年以上前にインストールしようとしたときはバグっぽい挙動で挫折した気がするけど、今度は比較的すんなり上手くいった。マッピング結果などを見るのがAccessで、マッピングやスキャッフォールディングを行うのがSolve。
BioNano Accessのインストール
公式のHPに従ってインストールすれば問題ない。https://bionano.com/wp-content/uploads/2023/02/CG-30170_Rev.AF_Bionano-Access-Installation-Guide.pdf
まず下記のソフトウェアをインストールし、
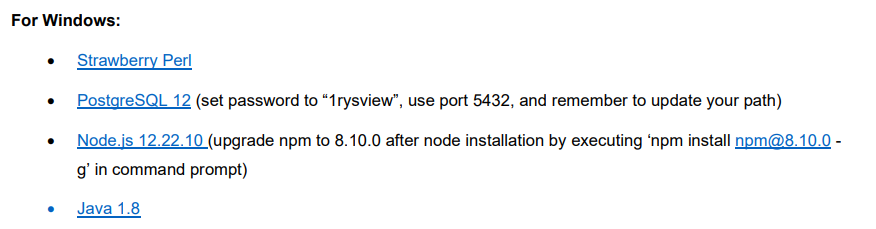
システム環境変数(PATH)に下記のようにPostgreSQLとJavaへのパスを追加。おそらくJavaのほうは細かいバージョンが違うパスになるはず。

システム環境変数の追加方法は下記ページ参照。https://atmarkit.itmedia.co.jp/ait/articles/1805/11/news035.html
ターミナルを開いて、下記を入力してBioNano Accessのウェブサーバを起動し、
cd c:\bionano\access\web\Server node --max-old-space-size=16384 server default
ウェブブラウザで http://localhost:3005/ を開く。ログインページが開いたら、下記のID, パスワードを入力。
Username: bionanoadmin Password: admin
BioNano Solveのインストール
基本的に公式HPの手順https://bionano.com/wp-content/uploads/2023/01/30182-Bionano-Tools-Installation-Guide.pdfに書いてある通りにやれば良い…はずなのだけど、書いていない前提が多い。まず、仮想マシンにCentOS7をインストールする。ユーザ名はbionanoで作ると想定されている感じがする。
次にCentOS7にグリッドエンジン( https://github.com/daimh/sge )を入れる。たぶん実際には使わないけど、インストールするときにqconfコマンドが使えないとエラーになる。具体的には下記の手順。
sudo su - yum groupinstall 'Development Tools' yum install git hwloc-devel openssl-devel libtirpc-devel motif-devel ncurses-devel libdb-devel pam-devel systemd-devel wget wget https://github.com/Kitware/CMake/releases/download/v3.23.0/cmake-3.23.0-linux-x86_64.tar.gz tar xvfz cmake-3.23.0-linux-x86_64.tar.gz export PATH=$(realpath cmake-3.23.0-linux-x86_64)/bin:$PATH git clone https://github.com/daimh/sge.git cd sge cmake -S . -B build -DCMAKE_INSTALL_PREFIX=/opt/sge cmake --build build -j sudo cmake --install build useradd -r -d /opt/sge sge chown -R sge /opt/sge cd /opt/sge yes "" | ./install_qmaster yes "" | ./install_execd source /opt/sge/default/common/settings.sh qhost -q #you should be able to see five lines of output qconf -as $HOSTNAME #add this node as submit host
そうすると、ようやくSolveのインストールマニュアルにある次のインストールが可能になる。
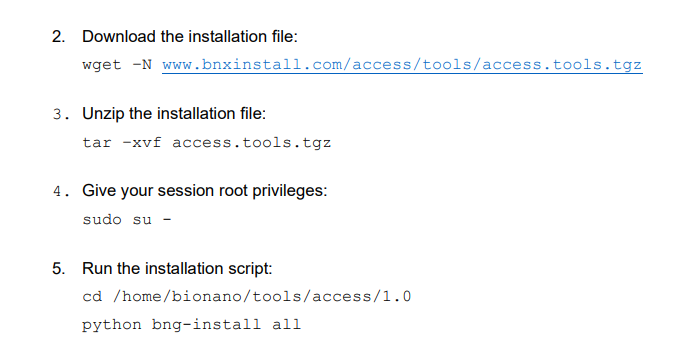
これでSolveの入ったDockerなどがインストールされる。AccessからSolveをGUIで呼び出せると良いのだけど、そもそも出来るかどうかわからなかった。コマンドラインからの使い方は公式HP (https://bionanogenomics.com/wp-content/uploads/2018/04/30205-Guidelines-for-Running-Bionano-Solve-Pipeline-on-Command-Line.pdf) にあるのだけど、コマンドラインから呼び出すにはDockerイメージを修正する必要がある。
具体的には下記のコマンドを入力する。
docker run -it --name solveinstall bionanodocker/bionano-docker-pipeline:latest bash cpanm install PerlIO::Util cpanm install Config::Simple cpanm install File::Slurp cpanm install File::Copy::Recursive cpanm install Fatal cpanm install XML::Simple cpanm install DateTime cpanm install DateTime::Format::Human::Duration cpanm install List::MoreUtils exit docker commit solveinstall mybionanosolve
その後、例えばNGSのscaffoldingを行うならば、下記のコマンドで実行できる。
docker run --user 1000:1000 --rm --privileged -v /home/bionano:/home/bionano -v $PWD:$PWD -w $PWD mybionanosolve perl /home/bionano/tools/pipeline/1.0/HybridScaffold/1.0/hybridScaffold.pl -n input_contigs.fasta -b EXP_REFINEFINAL1.cmap -u CTTAAG -c /home/bionano/tools/pipeline/1.0/HybridScaffold/1.0/hybridScaffold_DLE1_config.xml -r /home/bionano/tools/pipeline/1.0/RefAligner/1.0/RefAligner -o /home/bionano/output -f -g -B 2 -N 2 -p /home/bionano/tools/pipeline/1.0/Pipeline/1.0 -z results.zip -w status.txt # コンティグinput_contigs.fastaと、BioNanoでde novoスキャッフォールディングEXP_REFINEFINAL1.cmapを自分のファイルに変更
そもそもdocker使うなら、access.tools.tgzの中に入っているdockerイメージだけloadかimportしてしまえば、わざわざCentOS7を準備しなくてもよかった気はする。ここで解析した結果はAccessにインポートすると見られる。