本日の概要
先週の実験で、三四郎池、不忍池の水からDNAを抽出し、12S rRNAの全長(1,000 bp程度)、もしくは12S rRNAのMiFishプライマー領域(160-190 bp)をPCR増幅しました。また、池の水や発酵食品の「ヨーグルト」、「チーズ」、「イカの塩辛」、「生ハム」からから抽出したDNAを、16S rRNA全長(1,500 bp程度)をPCR増幅しました。本日は、Oxford Nanoporeという次世代シーケンサーで増幅したPCR産物をシーケンスしたデータを使って解析を行います。
本日のデータ解析のワークフローを以下に示します。
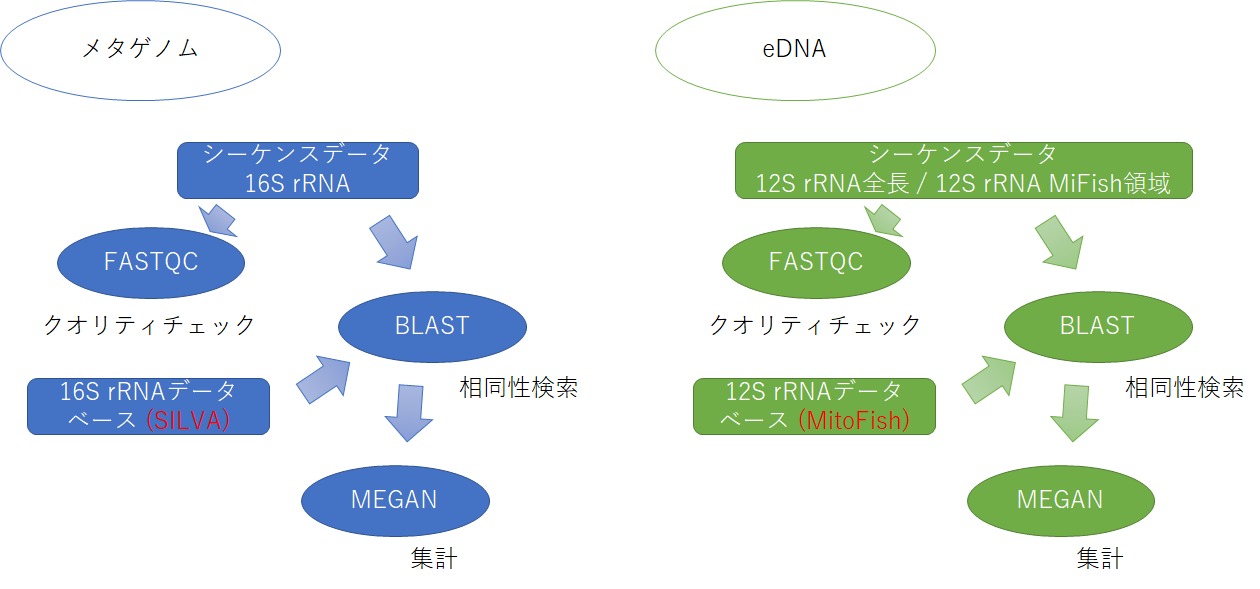
大雑把なデータ解析の流れとしては、 まずシーケンスデータの品質チェックをFastQCというツールを用いて行います。それから、手元のコンピュータにインストールしたBLASTを使って、すべてのシーケンスデータをデータベースと照合させ、各リードがどの種と相同性があるかを調べます。それからMEGANを使ってBLASTのヒットを集計し、各サンプルに含まれていたバクテリアの種類を網羅的に解析します。
環境DNAのサンプルは、MEGANの結果で検出された種と、実際に池にいそうな魚なのかの考察や、使用したプライマーの種類(12S全長 or 12S MiFish)の違いが結果に与える影響などを考えてみてください。メタゲノムのほうでは、食品のデータはシャッフルしてお渡ししますので、含まれるバクテリアの種類から、食品サンプルが上記4つの食品のどれであるかを推定します。また、池の水と食品で微生物叢にそれぞれ特徴があるかを考察してください。
準備( ツール・データのダウンロード )
BLASTのインストール
今回はメモリ8GBのWindowsノートPCを用います。NGSのデータ解析では往々にしてコマンドラインでの解析が必要であり、今回はBLASTの解析をコマンドラインで行います。まずは下記の手順でBLASTをセットアップしてください。
下記のようにエクスプローラでデスクトップを開いて、「ファイル」→「Windows PowerShellを開く」→「Windows PowerShellを開く」でPowerShellを開きます。
PowerShellが開いたら「wsl」と入力して、Windows Subsystem for Linux (WSL)を起動します。
WSLについての技術的な話は割愛しますが、上記手順でWindowsやMacとは別のOSであるUbuntu Linuxを起動することが出来ます。もし自分のWindowsコンピュータでWSLを実行したい場合は、こちらのページに手順を書いてありますので、参考にして下さい。WSLが起動したら、下記のように入力してください。
sudo apt update
#実行したら、パスワードを聞かれるので、「student」と入力します。入力しても何も表示されませんが、
#ちゃんと入力されていますので、「student」と入力して「Enter」キーを押してください。
sudo apt install -y ncbi-blast+
#↑でBLASTをインストールすることが出来ます。その他ツール・データベースのインストール
各班に1つ配布したUSB HDDに「day2\配布データ」というフォルダがあるので、それを「デスクトップ」等の適当なフォルダにコピーしておきます。もしくは個別に下記のファイルをすべてダウンロードしても良いです。
- FASTQC ・・・シーケンスデータのクオリティチェックを行う。公式HP
ダウンロードはここから - SILVA database ・・・16S rRNA (バクテリア・植物の葉緑体、合計約60万件)、18S rRNA (真核生物核ゲノム、約8万件)を網羅的に集めて整理したデータベース。ただし哺乳類の網羅率は高くはなく、哺乳類は407件登録されいてるが、そのうちヒトが162件。 公式HP
ダウンロードはここから - MitoFish ・・・魚類のミトコンドリアのデータベース。約2万種が登録されている。魚類ミトコンドリア上の16S rRNA、12S rRNAを探す場合に使用する。公式HP
ダウンロードはここから - MEGAN ・・・BLASTの結果から微生物叢情報や機能遺伝子情報へと変換してくれるソフトウェア。KEGGに関しては有償。公式HP
ダウンロードはここから - Nanoporeのシーケンスデータ ・・・メタゲノムとeDNAのデータが入っています。
ダウンロードはここから
Nanoporeシーケンスデータの説明
ナノポアでシーケンスを行ったPCR産物の電気泳動写真は下記の通り。

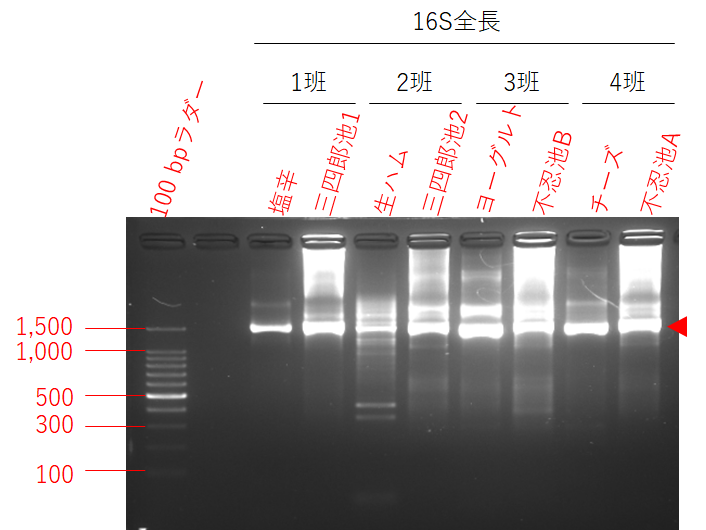
上記PCR産物を等濃度になるように混合してからナノポアのシーケンスを行っています。シーケンスデータのファイル名とサンプルの対応関係、取り出した配列の長さは下記の通り。
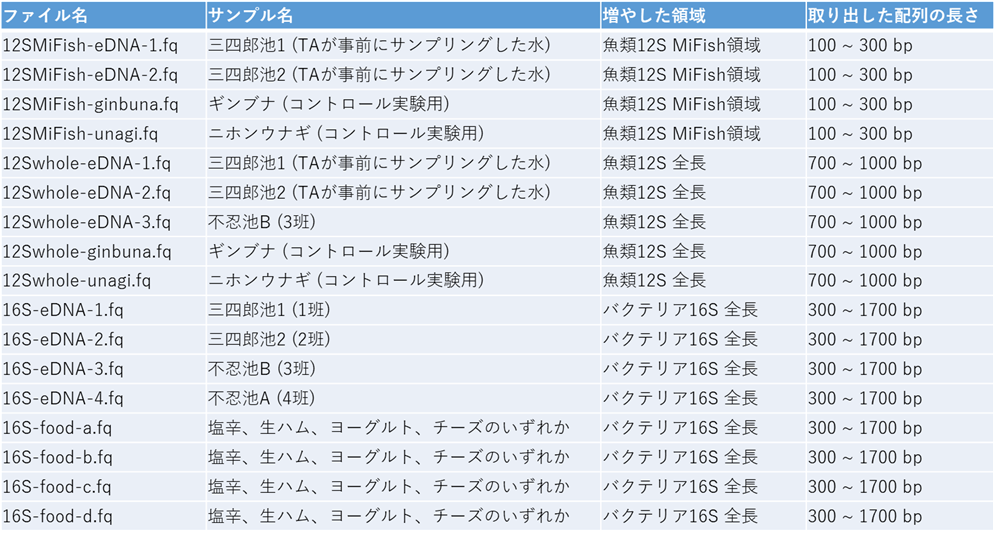
皆さんに渡したシーケンスデータは計算時間の関係で12Sは1,000リード、16Sは200リードだけにしていますが、各サンプルのリード数は本来下記のとおりです。 マルチプレックスタグをつけて1回のランで複数サンプルを同時にシーケンスしています。ナノポアのフローセルは再利用可能なので、2回目のランは1回目のフローセルを洗浄して使用しています。タグ間のコンタミや、1回目のランのデータが2回目のランにどの程度コンタミしているかは最後に考察課題としています。
ラン1回目
12SMiFish-eDNA-1 1472
12SMiFish-eDNA-2 1878
12SMiFish-unagi 77013
12SMiFish-ginbuna 36961
12Swhole-eDNA-1 2748
12Swhole-eDNA-2 8283
12Swhole-unagi 13459
12Swhole-ginbuna 7109
ラン2回目
12Swhole-eDNA-3 3312
16S-eDNA-1 25545
16S-eDNA-2 30731
16S-eDNA-3 22893
16S-eDNA-4 27453
16S-food-a 23794
16S-food-b 54185
16S-food-c 43172
16S-food-d 48692
ダウンロードしたツール・データの解凍
Explorerを開いて、ダウンロードフォルダに移動し、先ほどダウンロードした「fastqc_v0.11.8.zip」、「complete_partial_mitogenomes.zip」、「Megan_Community_x64_6.12.5.zip」、「SILVA_132_SSURef_Nr99_tax_silva.fasta.zip」、「sequence_data.zip」をダブルクリックして解凍します。 解凍されたファイルはデスクトップに保存されているはず。
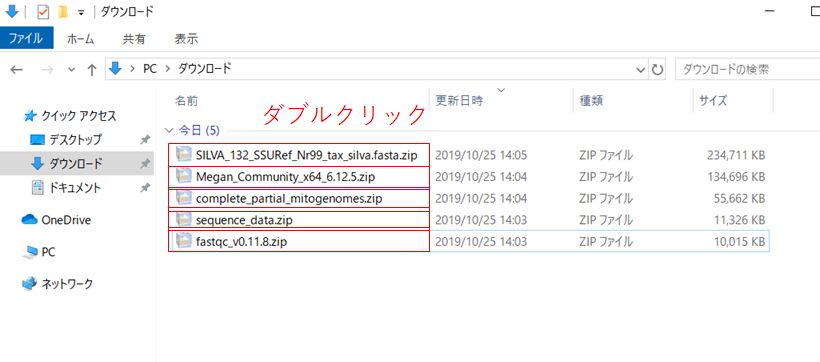
その中で、SILVA、MitoFishデータベースを解凍して出来たFASTAファイルや、ナノポアのシーケンスデータを解凍して出来たFASTQファイルはどこか一つのフォルダにまとめておいてください。
データ解析
FASTQC
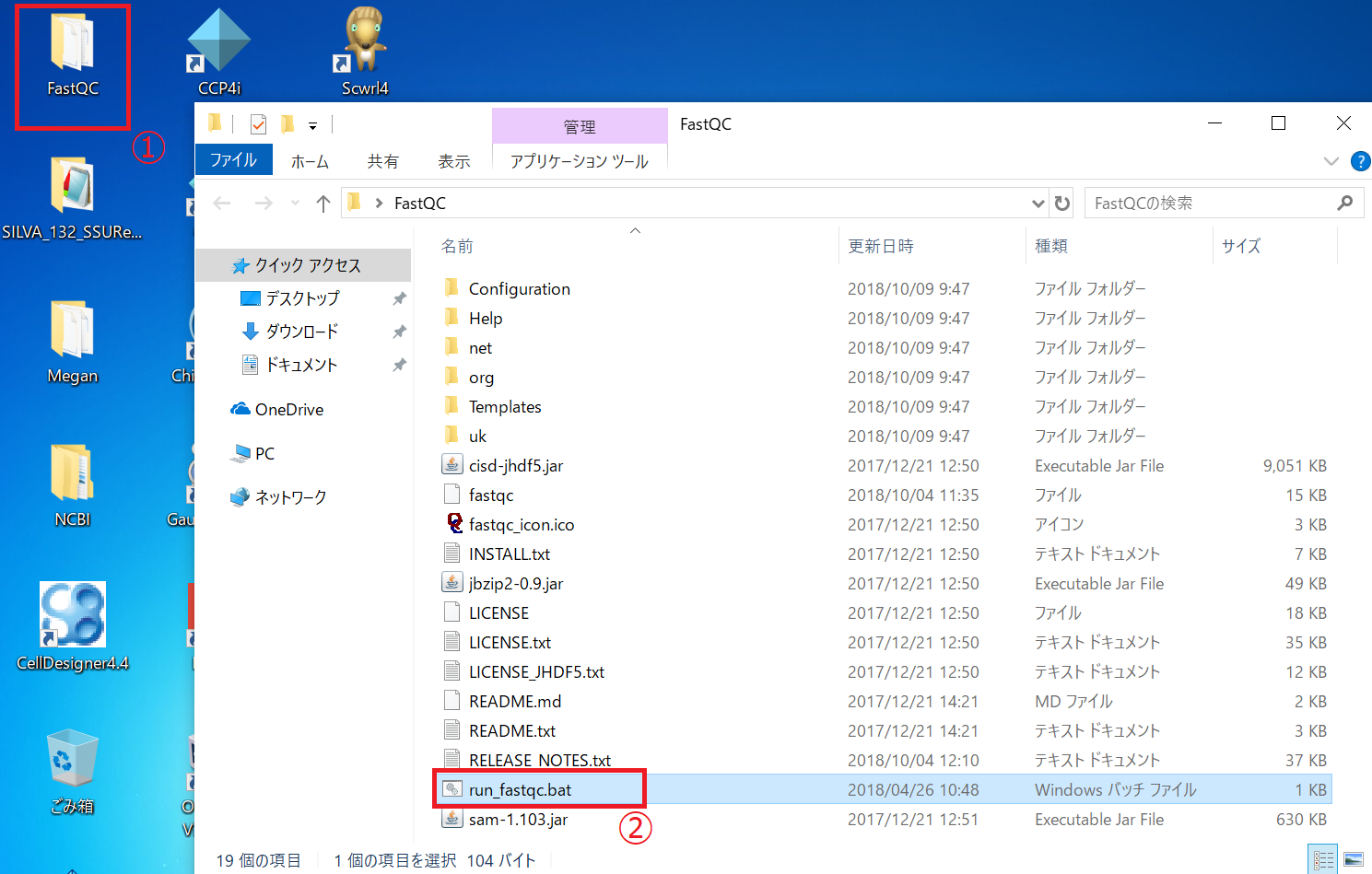
FastQCデスクトップにあるFastQCフォルダを開き、「run_fastqc.bat」をダブルクリックし、FastQCを実行します。
FastQCの上部メニューから、「File」→「Open」をクリックし、シーケンスデータ(FASTQファイル)を選択して開きます。選択ダイアログで「Shiftキー」もしくは「Ctrlキー」を押しながら複数ファイルをクリックすると、複数ファイルを選択できます。
{kind=link}
FastQCは主にIllumina用のクオリティチェックツールなので、Nanoporeのデータに対しては適切な評価ができておらず、評価値の〇×は気にしなくてよいです。下記はシーケンスデータのクオリティスコアに関する、平均値等の情報。
{kind=link}
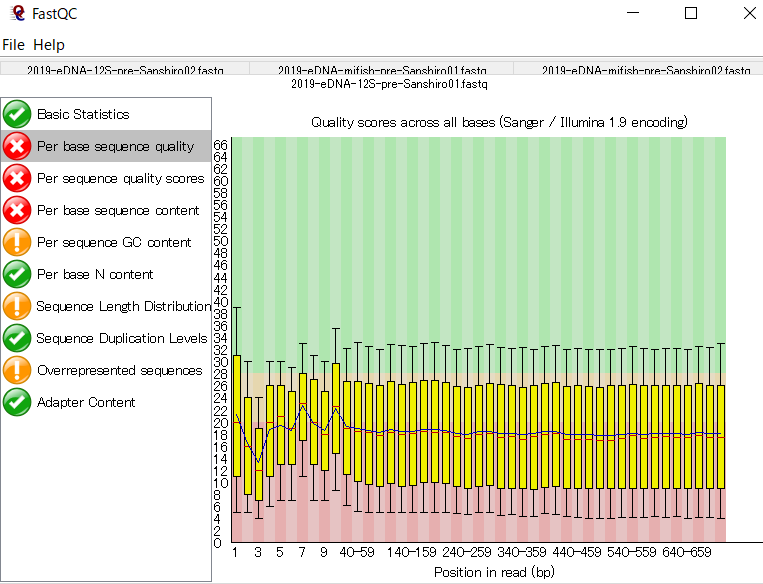
Nanoporeのデータだとクオリティスコア10強(精度90%強)となるはずである。
クオリティスコアQは、エラーの生じる確率 perror から下記のように計算されます。 (出典: https://bi.biopapyrus.jp/rnaseq/qc/fastq-quality-score.html )
{kind=link}
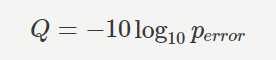
- クオリティスコアが 10 ならば、シーケンシングエラーが生じる確率は 10.0% であるから、読み取られた塩基の信頼度は 90.0% 。
- クオリティスコアが 20 ならば、シーケンシングエラーが生じる確率は 1.0% であるから、読み取られた塩基の信頼度は 99.0% 。
- クオリティスコアが 30 ならば、シーケンシングエラーが生じる確率は 0.1% であるから、読み取られた塩基の信頼度は 99.9% 。
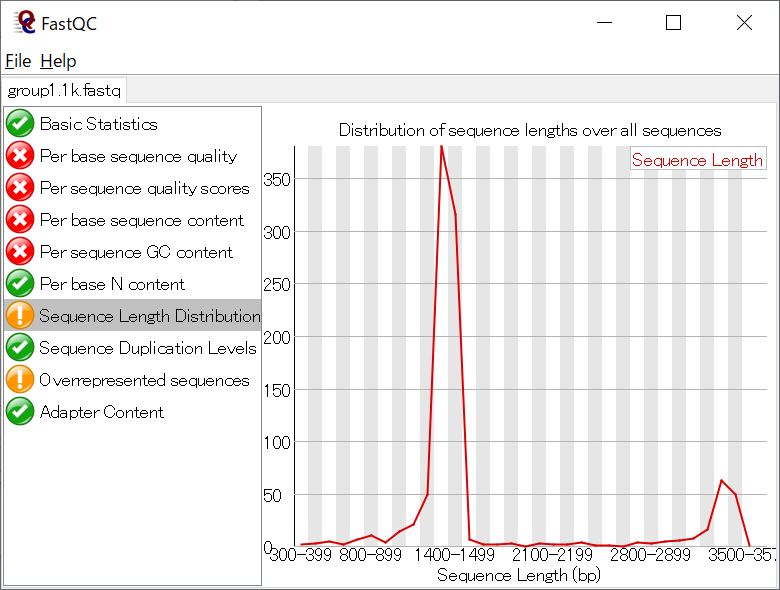
リード長の分布が想定される長さになっているかなども確認すること。 (16S全長は1.5 kbp程度、12S全長は1 kbp程度、12S MiFishは200 bp程度)
BLAST
FASTQ→FASTA変換
FASTQファイルをメモ帳などで開いてみると次のように表示されます。

FASTQ形式は 塩基配列とクオリティが両方含まれるファイル形式になりますが、BLASTのプログラムはFASTQ形式では入力ファイルとして受け付けてくれません。BLASTが対応しているのは、塩基配列だけの情報であるFASTA形式になります。
FASTA形式は1つのシーケンスにつき、「>」 で始まる1行のヘッダ行と、2行目以降のACGTのシーケンス文字列で構成されます。
(FASTAファイルの例)
>M12531.1 Human mitochondrial D-loop region with Phe-tRNA, 7S RNA, and 12S rRNA (5′ end) genes GAGCCGGAGCACCCTATGTCGCAGTATCTGTCTTTGATTCCTGCCTCATCCTATTATTTATCGCACCTACGTTCAATATTACAGGCGAACATACTTACTAAAGTGTGTTAATTAATTAATGCTTGTAGGACATAATAATAACAATTGAATGTCTGCACAGCCCCTTTCCACACAGACATCATAACAAAAAATTTCCACCAAACCCCCCCTCCCCCGCTTCTGGCCACAGCACTTAAACACATCTCTGCCAAACCCCAAAAACAAAGAACCCTAACACCAGCCTAACCAGATTTCAAATTTTATCTTTTGGCGGTATGCACTTTTAACAGTCACCCCCCAACTAACACATTATTTTCCCCTCCCACTCCCATACTACTAATCTCATCAATACAACCCCCGCCCATCCTACCCAGCACACACACCGCTGCTAACCCCATACCCCGAACCAACCAAACCCCAAAGACACCCCCCACAGTTTATGTAGCTTACCTCCTCAAAGCAATACACTGAAAATGTTTAGACGGGCTCACATCACCCCATAAACAAATAGGTTTGGTCCTAGCCTTTCTATTAGCTCTTAGTAAGATTACACATGCAAGC
そこで、まずFASTQのクオリティを削除して、FASTA形式に変換します。
- エクスプローラでシーケンスデータが入っているフォルダまで移動してから、PowerShellを開き、WSLを起動します。
- WSLの中で、下記のように一行で入力し、FASTQからFASTAへ変換します。
awk 'NR%4==1{print ">"substr($1,2)} NR%4==2{print $0}' input_file.fastq > output_file.fasta
# > output_file.fasta をつけると、出力をファイルに保存できる。
# input_file.fastq, output_file.fastaは適当に変更すること。 BLASTデータベース作成
SILVAデータベースのFASTAファイル、MitoFishデータベースのFASTAファイルから、makeblastdbコマンドによってBLASTのデータベースを作成します。WSLで下記のように入力します。
#SILVA
makeblastdb -in SILVA_132_SSURef_Nr99_tax_silva.fasta -dbtype nucl
#MitoFish
#都合により、ヒト・ブタのミトコンドリアも追加しておく
#ヒト・ブタのFASTAファイルダウンロードする。wgetはファイルをダウンロードするためのコマンド。
wget -O NC_000845.fasta "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nucleotide&id=NC_000845&rettype=fasta&retmode=text"
wget -O NC_012920.fasta "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nucleotide&id=NC_012920&rettype=fasta&retmode=text"
#MitoFishにヒト・ブタを追加する。catはファイルの中身を見るためのコマンド。
cat NC_000845.fasta NC_012920.fasta complete_partial_mitogenomes.fa > MitoFish-human-pig.fasta
makeblastdb -in MitoFish-human-pig.fasta -dbtype nucl-in には入力となるFASTAファイルを指定します。
-dbtype はFASTAファイルがDNA配列であれば「nucl」、アミノ酸配列であれば「prot」を指定します。
BLAST検索
FASTA形式に変換したシーケンスデータをクエリーとして、SILVAデータベースもしくはMitoFishデータベースで相同性検索を行います。WSLで次のように入力してください。
blastn -db SILVA_132_SSURef_Nr99_tax_silva.fasta -query input.fasta -num_threads 8 -out input.fasta.blastn-db には、BLASTデータベースファイルのprefixを指定します。今回の場合は元となったSILVAもしくはMitoFishのFASTAファイルを指定すれば良いです。
-query にはデータベースに対して相同性検索を行いたい問い合わせ配列が含まれるFASTAファイルを指定します。ここではFASTA形式に変換したシーケンスデータを指定します。(ファイル名は適宜変更すること。)
-num_threads には並列計算時に使用するCPUの数を指定します。演習で使用しているノートPCはCPUが8スレッドあるので、8を指定します。
-out には出力ファイルの名前を書く。(拡張子はMEGANに読み込ませるために「.blastn」とし、ファイル名は出来れば「入力FASTAファイル名」+「.blastn」としておくとMEGANがFASTAファイルを認識してくれて情報がリッチになる。)
BLAST結果例
下記のように入力すると、結果ファイルの中身を見ることが出来ます。結果表示を抜けるときは「q」を押します。
less input.fasta.blastn
# input.fasta.blastn は適当なファイル名に変更します。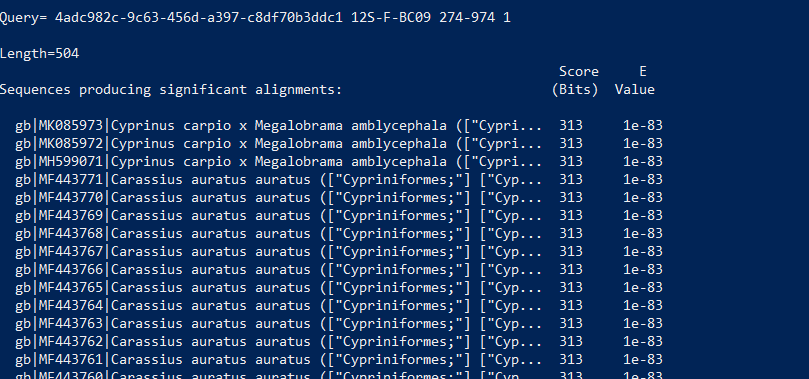
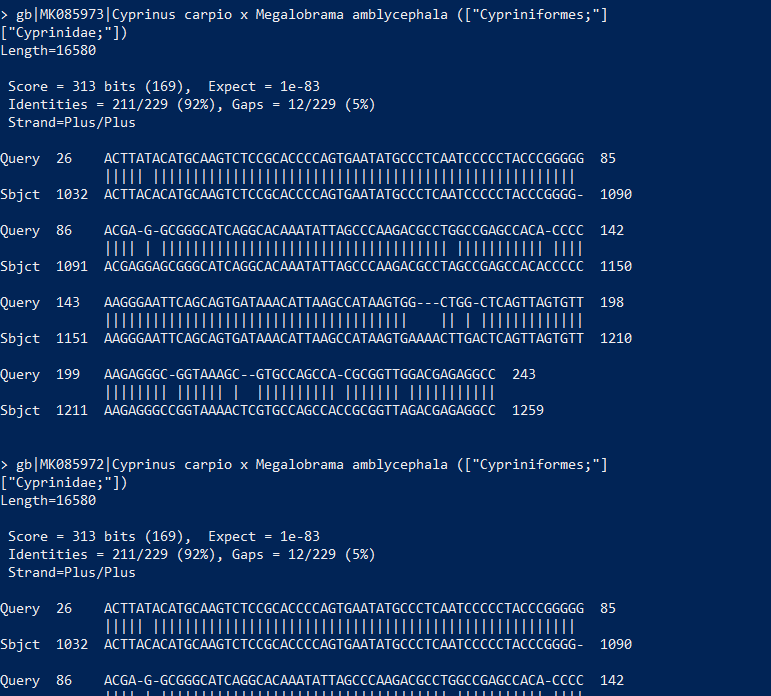
MEGANによるBLAST結果集計
MEGANではLowest Common Ancestor (LCA)法によってtaxon (分類群)を推測している。LCA法とは、1つのリードがデータベース中の複数の配列にヒットした場合、ヒットした配列に共通の分類群を求める方法です。 http://bio4j.com/blog/2012/02/finding-the-lowest-common-ancestor-of-a-set-of-ncbi-taxonomy-nodes-with-bio4j/
ただ、MEGANのデフォルト値はIlluminaシーケンサー用(精度99.9%)に設定されているので、ナノポアのように80~90%程度の精度では変更する必要があります。
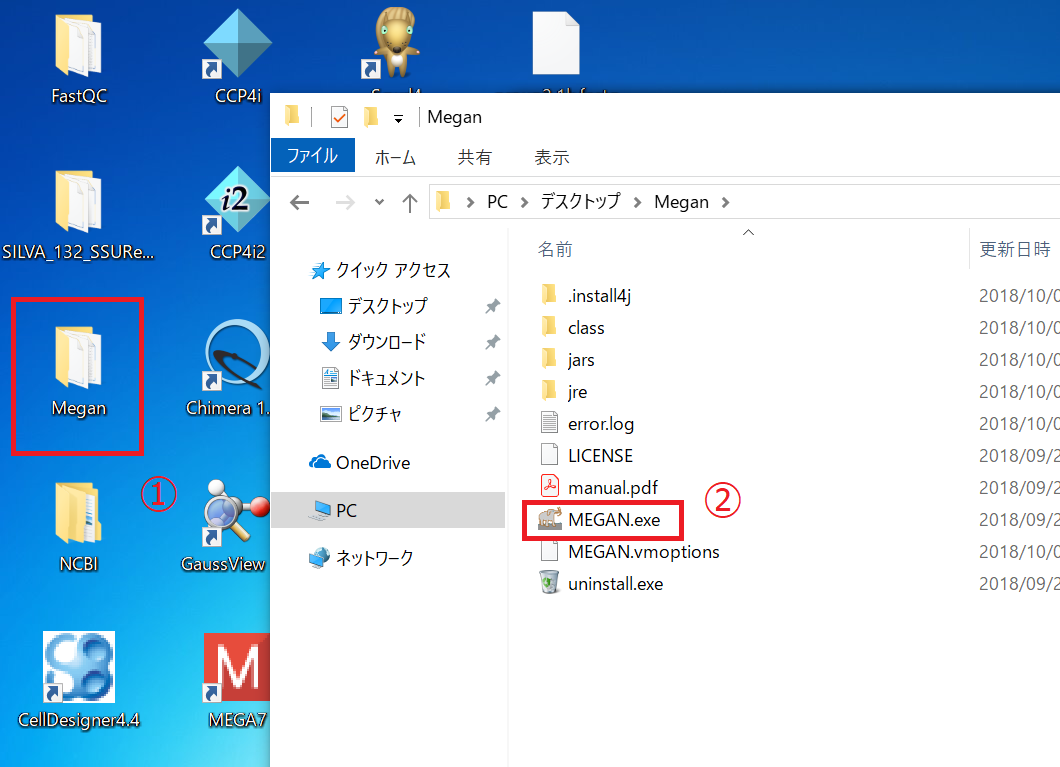
まずはMEGANを起動します。 「MEGAN.exe」をダブルクリックして起動してください。
MEGANが起動したら、「File」→「Import From BLAST」からBLAST結果を開きます。
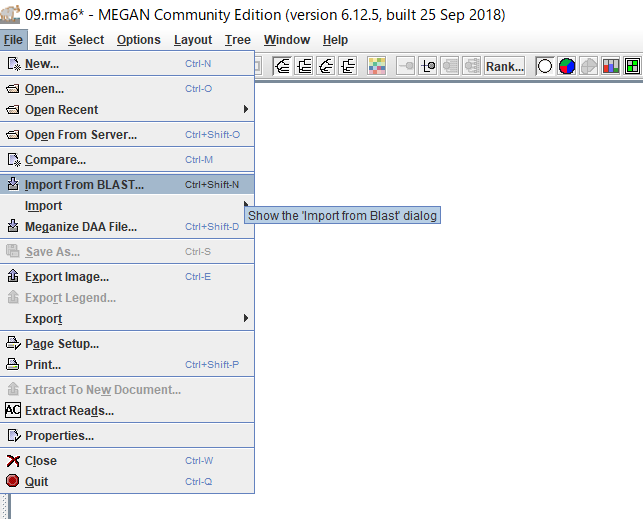
ダイアログ右上のボタンをクリックして、BLASTの結果ファイルを選択し、「Open」をクリックします。(複数ファイルを選択できますが、結果がマージされてしまうので、ファイルを一つだけ選択します。}
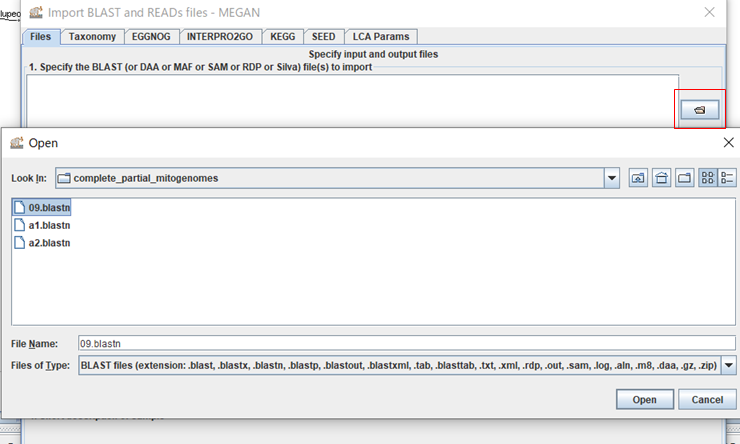
「LCA Params」タブを開いて、Top Percent: の値を0.5に変更しておきます。このパラメータは、BLASTの結果の中で最もスコアの高いトップヒットからどの程度離れたヒットまで使用するかの閾値になります。ナノポアではシーケンス精度が悪く、無関係な生物も似たようなスコアでヒットしてしまうため、ほぼトップヒットしか使わないように厳しめに閾値を設定しておきます。
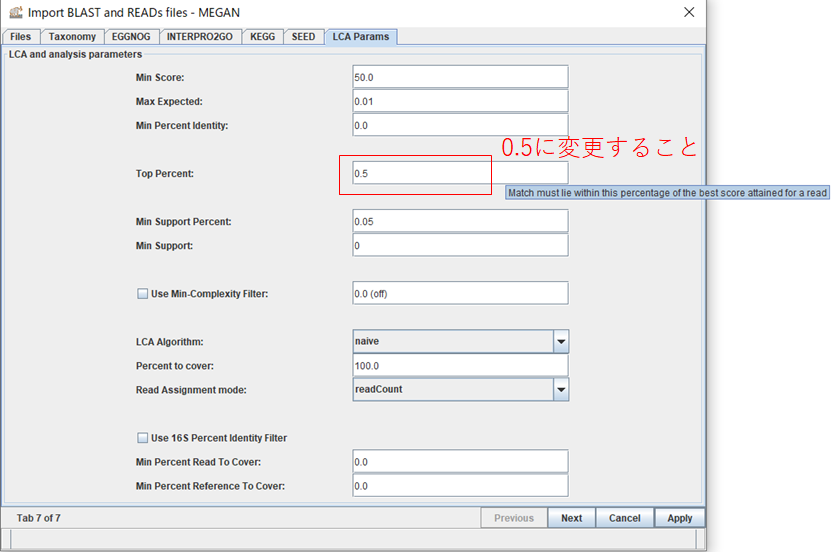
結果が表示されたら、 種名まで表示するために、「Rank」→「Species」を選択します。
↓
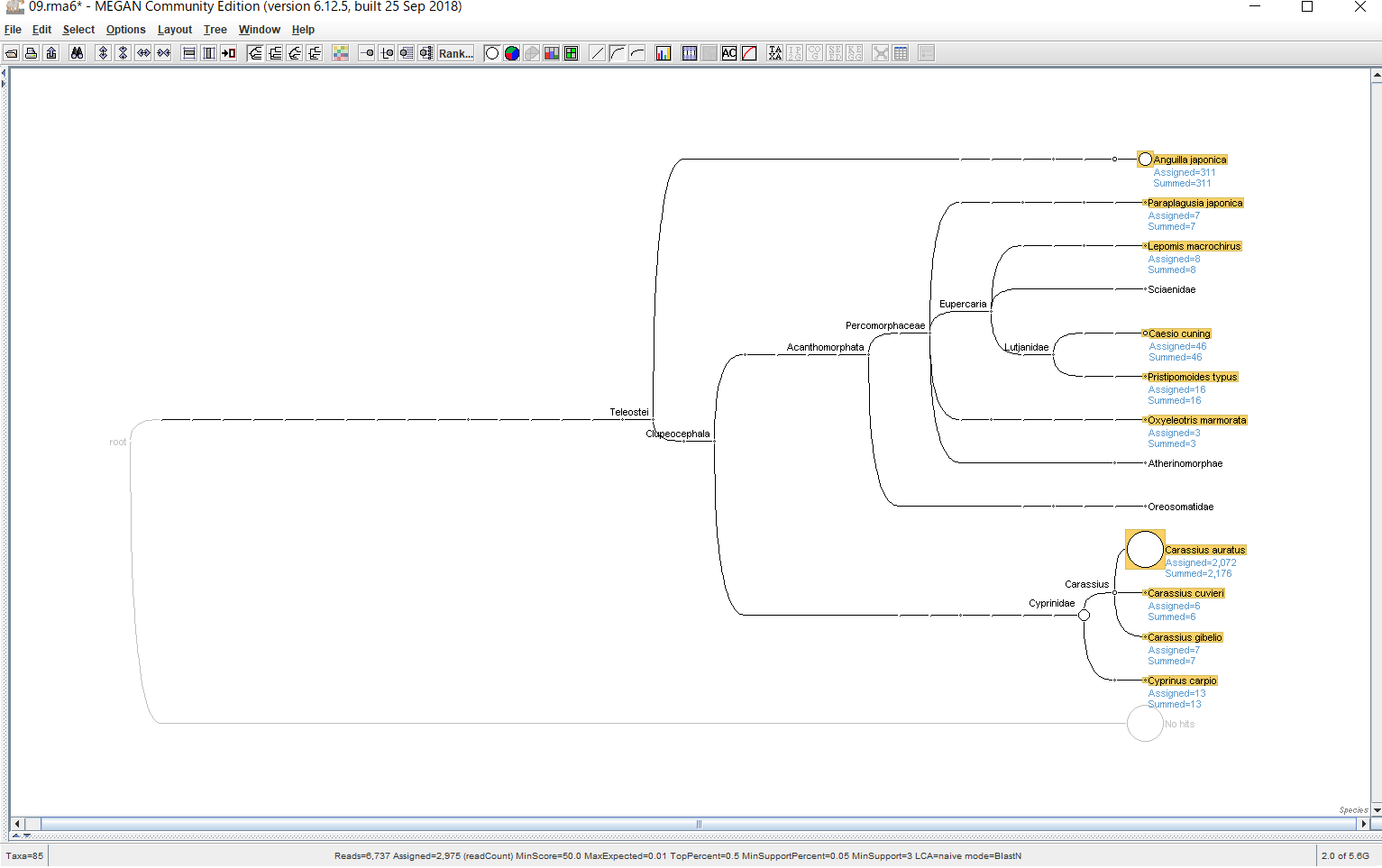
マウスホイールで拡大・縮小できます。(Let’s noteだと、マウスパッドの右端を上下になぞればスクロールになります。)
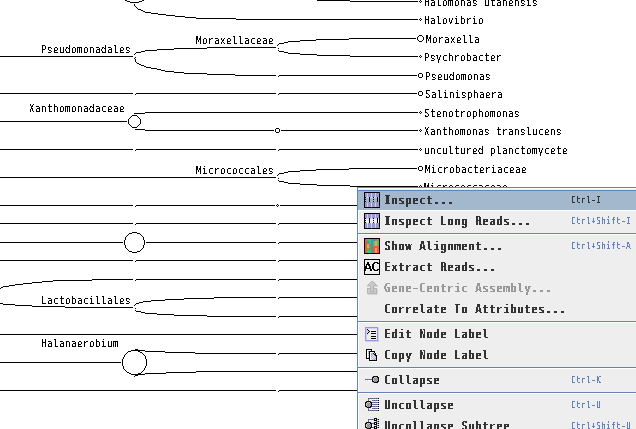
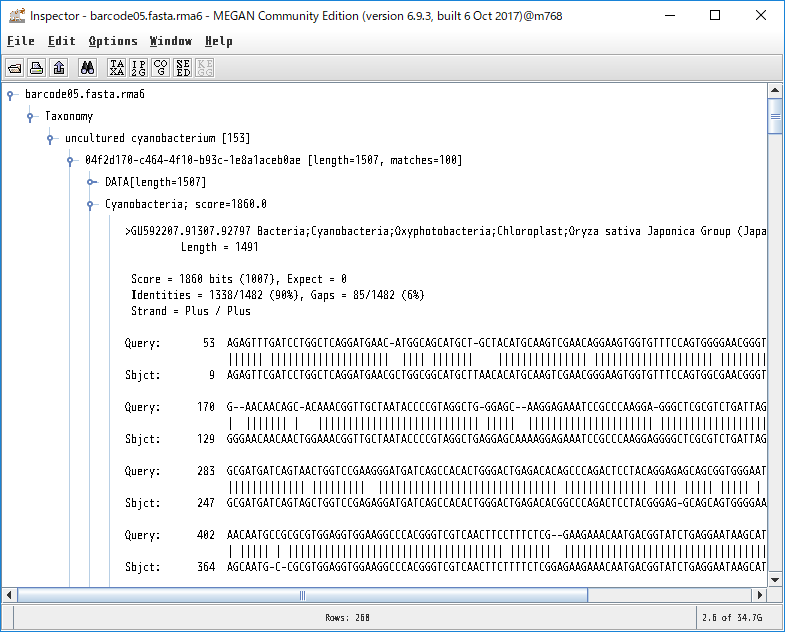
各生物種に割り当てられたBLASTの生データを見たい場合、ノードを右クリックし、「Inspector」を開けば見れます。本当にその生物はいるのか確認したくなった時などに、アライメント長や塩基配列そのものを確認するのに便利です。
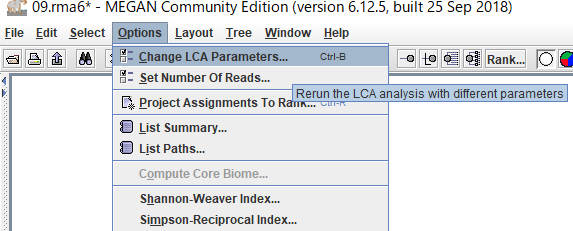
BLASTファイルを読み込むときに設定したLCAのパラメータを後から変更したい場合は、「Options」→「Change LCA Parameters」から可能です。
MEGANで複数サンプルを比較する場合は、blastnファイルを複数回開いたら、開いたウィンドウをそのままにした状態で、「File」→「Compare」とクリックして比較したいサンプルを選択し、「Apply」をクリックします。
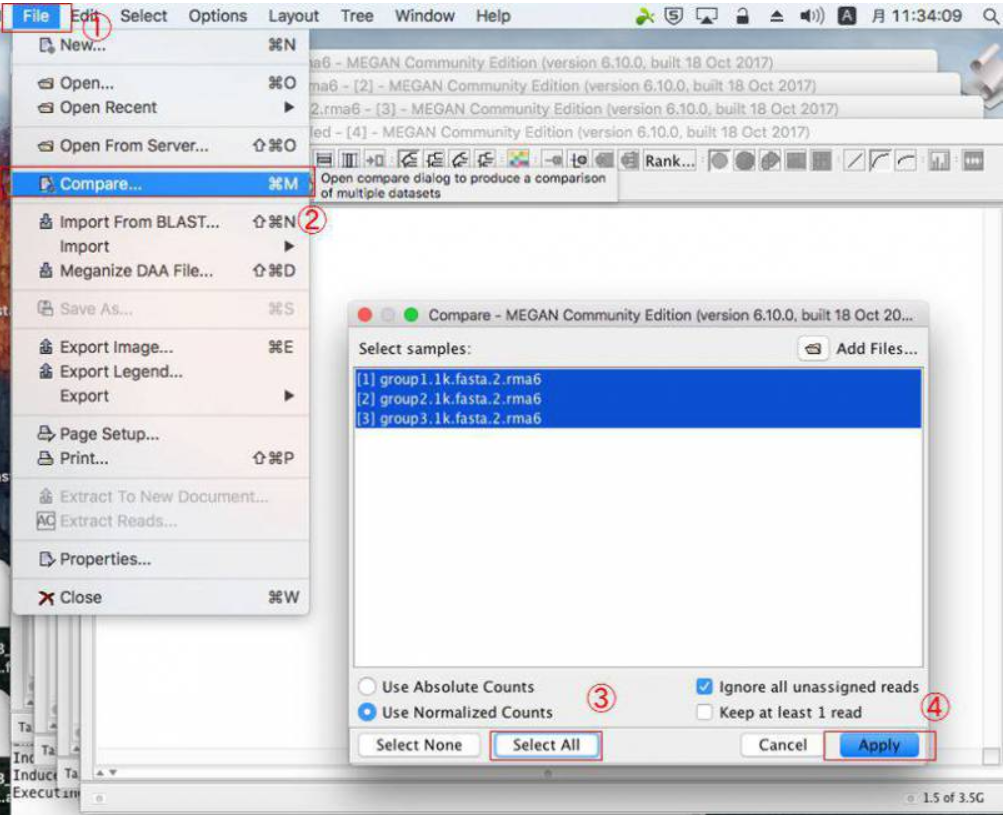
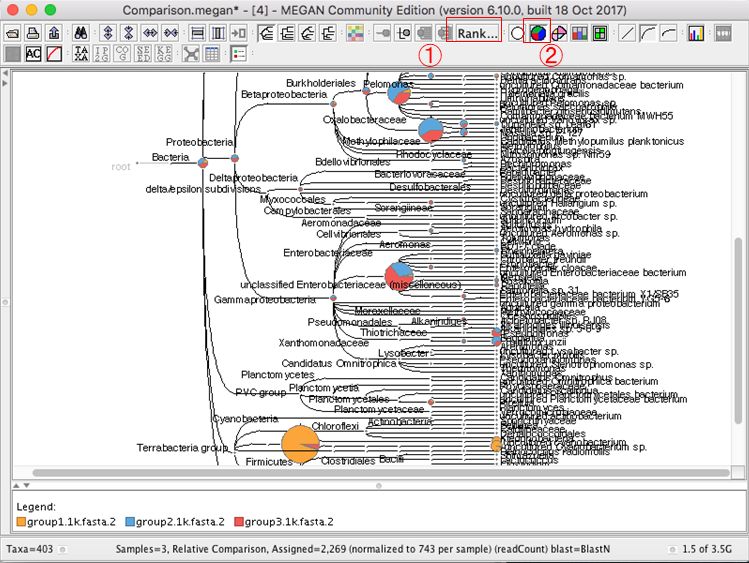
複数結果の表示を変更する場合、円グラフのボタンなどを押すと良いです。
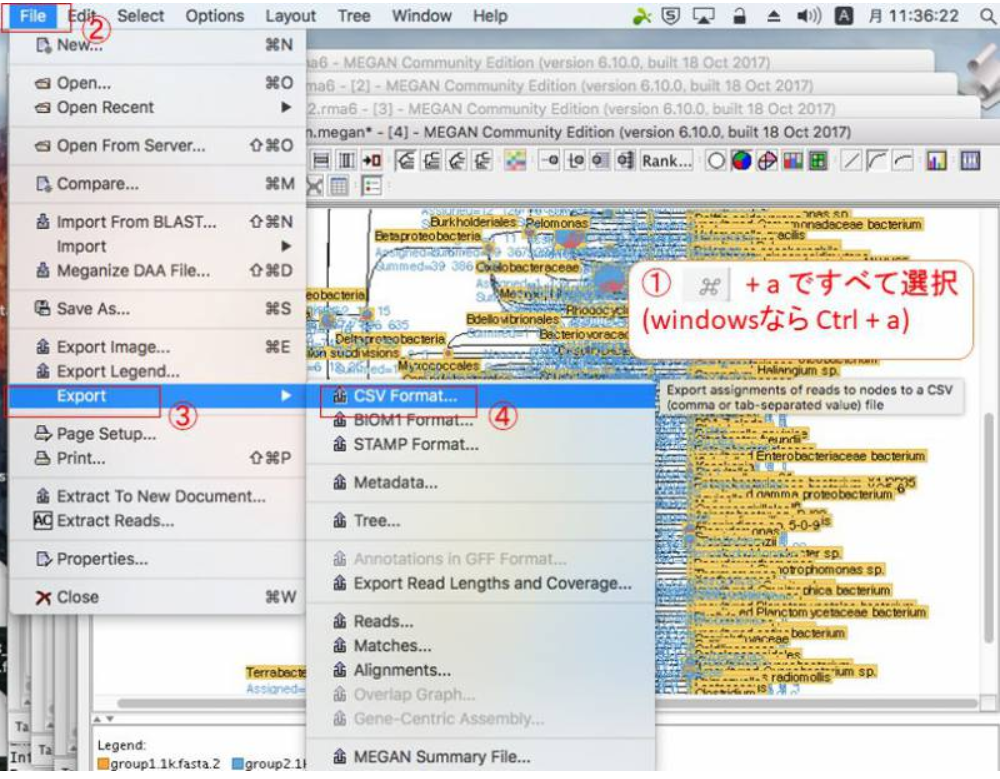
ExcelやR等で解析する場合は、データをタブ区切りテキスト形式でExportする必要があります。Exportしたいノードをクリックして(全部Exportする場合は全部選択(WidnowsならCtrl+a)して)、「File」→「Export」→「CSV Format」をクリックします。
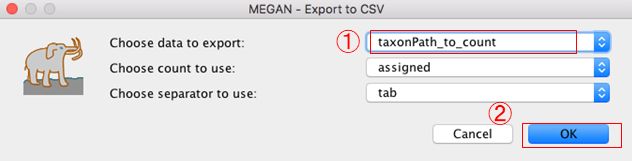
Exportするデータを「taxonPath_to_count」に変更します。(ほかのデータ形式でも勿論可)
適当な名前を付けて保存します。
Excelでの解析
ExportしたファイルをExcelで開くには、Excelを起動しておき、ExportしたファイルをExcel上にドラッグアンドドロップすれば良いです。
Excelでデータの概要を把握するのに役立つテクニックとして、条件付き書式を設定することで、データの大小を一目でわかるようにできたりします。
そのほか、「データ」→「フィルター」を使ってみたり、グラフを描いてみたりするのが通常の解析の流れになるかと思います。
課題
明日のプレゼンテーションに向けて、下記のテーマのいずれかを選び、手法、結果、考察を説明する資料をパワーポイントで作成すること。各班ごと4テーマで、必ず「食品からのメタゲノム解析」と「環境水からのeDNA解析 」は入れること。
- プラスミド抽出~制限酵素地図作成
- 魚肉からの魚種判定
- 食品からのメタゲノム解析
- 環境水からのメタゲノム解析
- 環境水からのeDNA解析
各テーマごとに下記の課題を考えてみること。インターネットを積極的に使用して調べることを推奨します。また、ある程度調べてもわからないことがあればTA・スタッフに聞いてみてください。
- プラスミド抽出~制限酵素地図作成
制限酵素処理がうまく行かなかった班が多かったかと思います。うまく行かなかった班は理由を考察してください。 - 魚肉からの魚種判定
昨日の実習では、MitoFishデータベースを使ったと思いますが、ほかにも有名なデータベースとしてNCBIのデータベース ( https://blast.ncbi.nlm.nih.gov/Blast.cgi )があります。こちらでもシーケンス結果を検索してみて、どの種にヒットするか、トップヒットした種はMitoFishデータベースと同じか調べてみてください。結果がNCBIとMitoFISHで同じでしょうか、違うでしょうか。どちらのデータベースのほうが良さそうでしょうか。 - 食品からのメタゲノム解析
food a ~ dは、 「イカの塩辛」、「生ハム」 、「ヨーグルト」、「チーズ」のどれかです。メタゲノムの結果からどの食品だったかを推測してください。理由も説明してください。 - 環境水からのメタゲノム解析
1年前の不忍池、三四郎池のメタゲノムのシーケンスデータが下記にあります。
http://suikou.fs.a.u-tokyo.ac.jp/yosh_data/2019jissyu/2018meta.zip
今年は台風のすぐあとの雨の日にサンプリングしましたが、1年前は通常の晴れの日でした。 1年前のデータと今年のデータを比べるとどのように違うでしょうか。その違いはなぜ生じたと考えられますか。昨年の実験プロトコール(実験テキスト)と今年のを比較して、プロトコールのどの部分が違うかも比べてみてください。 - 環境水からのeDNA解析
三四郎池と不忍池でeDNAから見つかる魚の種類は何種類?比較可能であれば、12S 全長と12S MiFish領域で差はみられるでしょうか。またどちらが実際の生物の種類・比率に近いでしょうか。
また、サンプルごとにタグをつけて一度にシーケンスしてから、シーケンス後でタグの配列を見てデータを分けていますが、間違って分類されてしまっているデータの割合はどの程度でしょうか? 12S全長、12S MiFishそれぞれで、ウナギやギンブナのコンタミの割合から推測してください。ちなみにIllumina社のシーケンサーでは0.1%程度タグを間違うことが知られています。
明日は3号館2階の講義室です。本日使用しているWiFiは使えませんので、各自U-Tokyo WiFiのIDを準備しておいてください。
提出物
明日の発表後に、作成した資料は班でまとめてHDDに入れて提出してください。