2018年 水圏生物科学実験III
本日の概要
先週の実験で、発酵食品の「ヨーグルト」、「納豆」、「イカの塩辛」、「ふぐの卵巣ぬか漬」、また池の水からDNAを抽出し、16S rDNA領域を増幅してOxford Nanoporeという次世代シーケンサーでシーケンスを行った。(食品のシーケンスデータはランダムにシャッフルして配布します。)
本日はナノポアのシーケンスデータを解析し、各シーケンスデータに含まれるバクテリアの種類を網羅的に解明します。
そして、含まれるバクテリアの種類から、食品サンプルが上記4つの食品のどれであるかを推定します。
また、池と食品で微生物叢に違いがあることを可視化します。
データ解析の流れとしては、まずシーケンスデータのチェックをFastQCというツールを用いて行い、その後で、rDNAのデータベースであるSILVA databaseに対して、シーケンスデータの相同性検索を行い、検索した結果をMEGANというツールで可視化し、統合する。
準備
ツール・データのダウンロード
各班に1つ配布したUSB HDDに「ツール、データベース、シーケンスデータ」というフォルダがあるので、それを「デスクトップ」等の適当なフォルダにコピーしておく。もしくは個別に下記のファイルをすべてダウンロードしても良い。
データの説明、用語解説
16S rDNAについて
学生実習で使用したプライマー
| 名前 | 配列 |
| 27F | AGAGTTTGATC(A/C)TGGCTCAG |
| 1492R | GGTTACCTTGTTACGACTT |
シーケンスデータの説明
シーケンスデータはFASTQというファイル形式で得られる
- FASTQ形式について
FASTQファイルをメモ帳などで開いてみると次のように表示される。

データベースの中から似ている塩基配列を探す方法について
■ 相同性検索について 東大 新領域 情報生命 笠原先生の講義資料より
まずは2つの配列がどの程度似ているのかをスコア化する。




■ 最も似ているアライメントを求めるSmith-Watermanアルゴリズム
https://en.wikipedia.org/wiki/Smith%E2%80%93Waterman_algorithm

■ 相同性検索を比較的高速に行うBLASTプログラム JST HPより
BLASTは、相同性検索(ホモ ロジーサーチ)を比較的高速に行うプログラムである。厳密な解を提供する Smith-Watermanアルゴリズムを少しヒューリスティックにすることで、完全な厳密解は与えないものの実用的には十分な精度を持ちつつ、 Smith-Watermanよりはるかに高速に検索を実現した。 また、BLASTではペアワイズの相同性検索の結果に対して、偶然そのような配列の一致が起こる期待値e-valueを出力し、閾値以上でデータベースとヒットした結果を出力する。
BLASTでは、問い合わせ配列とデータベース配列の組み合わせから、次の5種類が用意されている。
| プログラム名 | query | db | |
| blastn | DNA | DNA | |
| blastp | protein | protein | |
| blastx | DNA | protein | (DNAはアミノ酸に翻訳して比較) |
| tblastn | protein | DNA | (DNAはアミノ酸に翻訳して比較) |
| tblastx | DNA | DNA | (DNAはアミノ酸に翻訳して比較) |
■ Blastの結果についての説明 https://www.jaici.or.jp/stn/pdf/seqfaq.pdf

データ解析
解析は次の流れで行う。
- FastQCでシーケンスデータ(FASTQファイル)のリード数、平均リード長、クオリティ等を確認
- FASTQをFASTAファイルに変換
- BLASTを使用し、変換したFASTAファイルに相同な配列をSILVAデータベースから検索する。
- BLASTの結果ファイルをMEGANで読み込む。
- Excelでグラフ化する
ダウンロードしたツール・データの解凍
- ダウンロードしたファイルの解凍
Explorerを開いて、ダウンロードフォルダに移動し、先ほどダウンロードした
「fastqc_v0.11.8.zip」、「blast-2.7.1.zip」、「Megan_Community_x64_6.12.5.zip」、「SILVA_132_SSURef_Nr99_tax_silva.fasta.zip」をダブルクリックして解凍する。 解凍されたファイルはデスクトップに保存されているはず。(もしデスクトップに解凍されていなければ、ファイルをデスクトップに移動しておいてください。)
- シーケンスデータをデスクトップで解凍
ツールと同様にシーケンスデータ「seq.zip」をダブルクリックすると、デスクトップにシーケンスデータが解凍される。(もしデスクトップに解凍されていなければ、ファイルをデスクトップに移動しておいてください。)
- FastQC
デスクトップにあるFastQCフォルダを開き、
「run_fastqc.bat」をダブルクリックし、FastQCを実行する。
FastQCの上部メニューから、「File」→「Open」をクリックし、シーケンスデータ(FASTQファイル)を選択して開く。
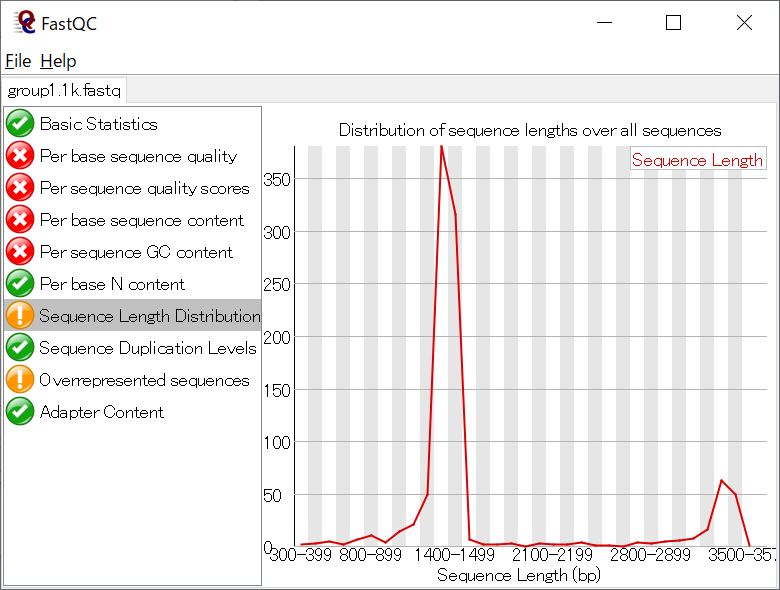
FastQCは主にIllumina用のクオリティチェックツールであり、Nanoporeのデータに対しては適切な評価ができないので、評価値の〇×は気にしなくてよい。下記はシーケンスデータのクオリティスコアに関する、平均値等の情報。クオリティスコアに関する説明はこちら。Nanoporeのデータだとクオリティスコア10強(精度90%強)となるはずである。
リード長の分布がPCRで増幅した長さになっているかなども確認すること。




4.FASTQ→FASTA変換
FASTA形式は1つのシーケンスにつき、「>」 で始まる1行のヘッダ行と、2行目以降の実際のシーケンス文字列で構成される。BLASTのプログラムではFASTQ形式では入力ファイルとして受け付けてくれないため、FASTQ形式のクオリティを削除したFASTA形式に変換する必要がある。
(FASTAファイルの例) >gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus] LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLV EWIWGGFSVDKATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLG LLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIVIL GLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGX IENY
まずExplorerでデスクトップを開いておく。

Explorerの上部メニューから、「ファイル」→「Windows PowerShellを開く」→「Windows PowerShellを開く」をクリックして、PowerShellを起動する。

PowerShellのようなターミナルでよく使用するコマンドを幾つか挙げておく。
ls : 現在のディレクトリ内のファイルを一覧表示。
cd [ディレクトリ名] : 指定したディレクトリに移動する。
more [ファイル名] : 指定したファイルの中身を表示する。表示をやめるときは「q」を押す。
下記のコマンドを入力し、FASTQファイルからクオリティを除去したFASTAファイルへと変換する。
・FASTQからFASTAへ変換する関数の作成 (下記をコピーして、PowerShellに張り付けること。PowerShell上で右クリックすれば貼り付け可能。)
function fastq_to_fasta(){
begin{$n=1}
process{if($n%4 -eq 1){$_ -replace "^@", ">"}elseif($n%4 -eq 2){$_}; $n+=1}
}
・FASTQ→FASTA変換 (入力ファイル「group1.1k.fastq」、出力ファイル「group1.1k.fasta」の名前は各自適当に変更すること)
cat group1.1k.fastq|fastq_to_fasta |Out-File -Encoding ascii -FilePath group1.1k.fasta

5. BLASTデータベース作成
SILVAのrDNAの配列がすべて含まれるFASTAファイルから、makeblastdbコマンドによってBLASTのデータベースを作成する。(PowerShellで下記のように入力する。)
NCBI\blast-2.7.1+\bin\makeblastdb.exe -in SILVA_132_SSURef_Nr99_tax_silva.fasta\SILVA_132_SSURef_Nr99_tax_silva.fasta -dbtype nucl
-in には入力となるFASTAファイルを指定する。
-dbtype はFASTAファイルがDNA配列であれば「nucl」、アミノ酸配列であれば「prot」を指定する。
6. BLAST検索
FASTA形式に変換したシーケンスデータをクエリーとして、SILVAデータベースに塩基配列の相同性検索を行う。(PowerShellで作業する。)
NCBI\blast-2.7.1+\bin\blastn.exe -db SILVA_132_SSURef_Nr99_tax_silva.fasta\SILVA_132_SSURef_Nr99_tax_silva.fasta -query group1.1k.fasta -num_threads 4 -out group1.1k.fasta.blastn
-db には、BLASTデータベースファイルのprefixを指定する。今回の場合は元となったFASTAファイルを指定すればよい。
-query にはデータベースに対して相同性検索を行いたい問い合わせ配列が含まれるFASTAファイルを指定する。ここではFASTA形式に変換したシーケンスデータを指定する。(各班のファイルを適宜指定すること。)
-num_threads には並列計算時に使用するCPUの数を指定する。演習で使用しているノートPCはCPUが4コアあるので、4を指定。
-out には出力ファイルの名前を書く。(拡張子はMEGANに読み込ませるために「.blastn」とすること)
7. MEGANによる結果表示
MEGANではLowest Common Ancestor (LCA)法によってtaxon (分類群)を推測している。LCA法とは、1つのリードがデータベース中の複数の配列にヒットした場合、ヒットした配列に共通の分類群を求める方法である。http://bio4j.com/blog/2012/02/finding-the-lowest-common-ancestor-of-a-set-of-ncbi-taxonomy-nodes-with-bio4j/

まずはMEGANを起動する。

次にBLAST結果ファイルをMEGANで開く。(以下はMacでの操作画面のキャプチャであるが基本的には同じである。)



LCAのパラメータを変更する場合は、ここで変更する。(後からでも変更可能)

結果が表示される。

種名まで表示するために、表示するRankを変更する。


あるtaxonに割り当てられた生データを見たい場合、ノードを右クリックし、「Inspector」を開けば良い。

Inspectorでは、該当taxonに割り当てられたリードと、blastの結果も全て表示させることが可能。

6.MEGANで複数サンプルの比較
blastnファイルを複数回開いたら、開いたウィンドウをそのままにした状態で、「File」→「Compare」とクリックして比較したいサンプルを選択し、「Apply」をクリックする。

比較ウィンドウを開くと、とりあえず下記のように表示される。

表示形式を変更したりすると下記のように表示される。

ExcelやR等で解析する場合は、データをタブ区切りテキスト形式でExportする。Exportしたいノードをクリックして(全部Exportする場合は全部選択して)、「File」→「Export」→「CSV Format」をクリックする。

Exportするデータを「taxonPathtocount」に変更してみる。(ほかのデータでも勿論可)


7.Excelでの解析
ExportしたファイルをExcelで開くには、Excelを起動しておき、ExportしたファイルをExcel上にドラッグアンドドロップすればよい。

フィルターを使ってみたり、グラフを描いてみたりする。Excelでデータの概要を把握するのに役立つテクニックとして、条件付き書式を設定することで、データの大小を一目でわかるようにできたりする。

問題
- food1 ~ 4は、「ヨーグルト」、「納豆」、「イカの塩辛」、「ふぐの卵巣ぬか漬」のどれか。理由も説明せよ。
- 自分たちの班の池のシーケンスデータのリード数、最長リード長、最短リード長はいくつか。
- 池の水の結果を比較し、考察せよ。
提出物
下記のファイルは明後日皆さんに行って頂く研究発表で使用するので、USB HDDに入れて提出してください。
- 池の水を採取した時の写真
- Blast結果、MEGANの結果、Excelでまとめたファイルなど