**文書の過去の版を表示しています。**
メタゲノム・eDNA解析 2024
先週の実験で、三四郎池の水や様々な食品からDNAを抽出し、PCR増幅しました。本日は、Oxford Nanoporeという次世代シーケンサーで増幅したPCR産物をシーケンスしたデータを使って解析を行います。
PCR増幅したサンプルリスト
| サンプル | プライマー領域 | 対象生物 | PCR断片長 |
| 三四郎池の水 | ミトコンドリア12S rRNA | 魚 | 200 bp強 |
| 三四郎池の水 | バクテリア16S rRNA | バクテリア | 1,500 bp程度 |
| 発酵食品 | バクテリア16S rRNA | バクテリア | 1,500 bp程度 |
本日のデータ解析のワークフローを以下に示します。

大雑把なデータ解析の流れとしては、 まずシーケンスデータの品質チェックをFastQCというツールを用いて行います。それから、コンピュータにインストールしたBLASTを使って、すべてのシーケンスデータをデータベースと照合させ、各リードがどの種と相同性があるかを調べます。それからMEGANを使ってBLASTのヒットを集計し、各サンプルに含まれていたバクテリアの種類を網羅的に解析します。
サンプルの説明
三四郎池で採水したときの様子
1班

2班

3班

4班

発酵食品
1班:ヨーグルト
2班:マッコリ
3班:ナンプラー
4班:チーズ
PCR結果まとめ
1班:

2班:

3班:

4班:

スタッフによる追加実験:

ナノポアシーケンス結果
クオリティスコアQから、エラーの生じる確率 P(error) は下記のように計算されます。 (出典: https://bi.biopapyrus.jp/rnaseq/qc/fastq-quality-score.html )

- クオリティスコアが 10 ならば、シーケンシングエラーが生じる確率は 10.0% であるから、読み取られた塩基の信頼度は 90.0% 。
- クオリティスコアが 20 ならば、シーケンシングエラーが生じる確率は 1.0% であるから、読み取られた塩基の信頼度は 99.0% 。
- クオリティスコアが 30 ならば、シーケンシングエラーが生じる確率は 0.1% であるから、読み取られた塩基の信頼度は 99.9% 。
A. 準備( ツール・データのダウンロード )
1. ツールのインストール
これからダウンロードするファイルを入れるディレクトリを作成し、その中にすべてのファイルをダウンロードします。とりあえずここではダウンロードフォルダの中に「2024jissyu」というフォルダを作り、そこにこれからダウンロードするファイルを全部移動させたことを前提に書いてあります。(注意:今回使用するプログラムはフォルダ名が日本語になっていると正常動作しません。もしWindowsで「C:\Users\XXX\Downloads\東大学生実験\2024jissyu」などとパスに日本語が含まれる場合は、「C:\Users\XXX\Downloads\2024jissyu」など途中に日本語フォルダを挟まないフォルダを作ってその中にダウンロードして下さい。OneDriveでダウンロードフォルダを同期させていると「C:\Users\XXX\ダウンロード」と日本語になっていることもあるので注意。)
- SeqKit
https://bioinf.shenwei.me/seqkit/download/
Windowsは「Windows amd64」の「seqkit_windows_amd64.exe.tar.gz」、Mac (Intel)は「seqkit_darwin_amd64.tar.gz」、Mac (2020年のM1プロセッサ以降)は「seqkit_darwin_arm64.tar.gz」をダウンロードする。
WindowsではスタートメニューからPowerShellを起動してターミナルを開く。MacではLaunchpad→その他→ターミナルを起動する。
ターミナルに下記のコマンドを入力し、seqkitを解凍する。
#ダウンロードしたディレクトリに移動する。 cd ~/Downloads/2024jissyu/ #ダウンロードしたファイルを解凍する。(Win) tar vxf seqkit_windows_amd64.exe.tar.gz #ダウンロードしたファイルを解凍する。(Mac) tar vxf seqkit_darwin_*.tar.gz
- BLAST
https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/2.16.0/
Windowsは「ncbi-blast-2.16.0+-x64-win64.tar.gz」を、Macは「ncbi-blast-2.16.0+-universal-macosx.tar.gz」をダウンロードし、「2024jissyu」フォルダに移した後で、下記のコマンドを実行する。
#ダウンロードしたファイルを解凍する。 ## Windows tar vxf ncbi-blast-2.16.0+-x64-win64.tar.gz ## Mac tar vxf ncbi-blast-2.16.0+-universal-macosx.tar.gz
- FastQC
https://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc
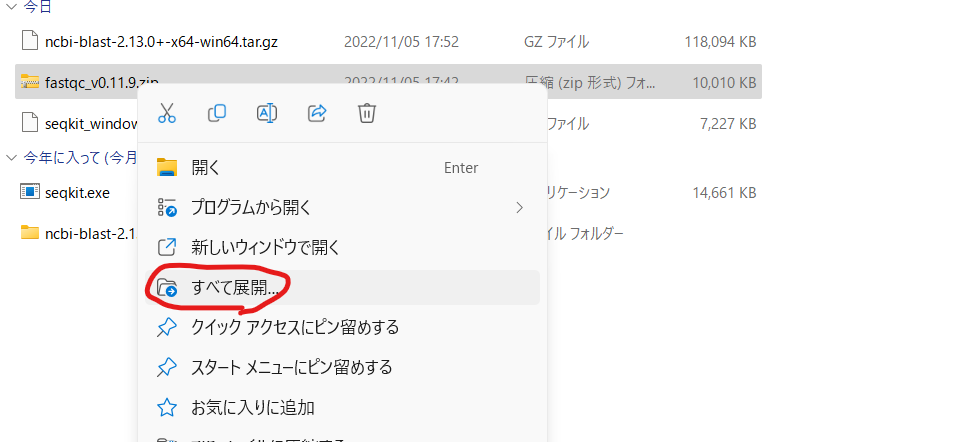
Windowsは「FastQC v0.12.1 (Win/Linux zip file)」をダウンロードし、上図のように解凍するか、もしくは「2024jissyu」フォルダに移した後で、下記のコマンドを実行する。

Expand-Archive -Path fastqc_v0.12.1.zip -DestinationPath .
Macは「FastQC v0.12.1 (Mac DMG image)」をダウンロードし、ダブルクリックしてイメージをマウントしておく。
- Javaランタイム
FastQCを実行するのに必要。https://www.java.com/ja/download/manual.jsp より適切なJavaをインストールする。
Windowsは「jre-8u431-windows-x64.exe」, MacはIntel系のCPUは「jre-8u431-macosx-x64.dmg」を、M1以降のCPUは「jre-8u431-macosx-aarch64.dmg」のインストーラーをダウンロードし、実行してインストールする。

- MEGAN
http://software-ab.cs.uni-tuebingen.de/download/megan6/welcome.html
Windowsは「MEGAN6 Community Edition installers」の「MEGAN_Community_windows-x64_6_25_10.exe」をダウンロードし、ダウンロードしたファイルをクリックorダブルクリックしてインストーラーを起動する。

下記の画面が出たら「詳細情報」を開いて

「実行」をクリックする。

インストーラーに従い、「次へ」等を押してインストールを完了する。

Macは「MEGAN_Community_macos_6_25_10.dmg」をダウンロードし、ダウンロードしたファイルをダブルクリックしてディスクイメージをマウントし、インストーラーをダブルクリックして起動する。

開いて良いか聞かれたら「開く」を押す。

インストーラーに従い、「次へ」等を押してインストールを完了する。
2. データベースのダウンロード
- 16S rRNA、18S rRNA、ミトコンドリア、葉緑体等のメタバーコーディングで使用される領域をまとめたFASTAファイルをダウンロードして各自OSの機能でzipファイルを解凍し、中身の
silva-SSU-LSU_PR2_NCBI-16S-mito-plastid_2023-04-18_rename_mitofish-2023-11-07.fastaを「2024jissyu」フォルダにコピーしておく。
このファイルは、NCBI blast database (ミトコンドリア、葉緑体)、PR2(18S rRNAなど)、SILVA(16S rRNA, 23S rRNA)、MitoFish (MiFish用12S rRNA)をマージして作ったもの。具体的には下記のファイルをマージしたもの。
https://ftp.ncbi.nlm.nih.gov/blast/db/ から16S_ribosomal_RNA、LSU_prokaryote_rRNA、mito
http://ftp.ncbi.nih.gov/refseq/release/plastid/ からplastid.*.genomic.fna.gz
https://github.com/pr2database/pr2database/releases/download/v4.14.0/pr2_version_4.14.0_SSU_taxo_long.fasta.gz
https://www.arb-silva.de/fileadmin/silva_databases/current/Exports/SILVA_138.1_LSURef_NR99_tax_silva_trunc.fasta.gz
https://www.arb-silva.de/fileadmin/silva_databases/current/Exports/SILVA_138.1_SSURef_NR99_tax_silva_trunc.fasta.gz
http://mitofish.aori.u-tokyo.ac.jp/species/detail/download/?filename=download%2F/complete_partial_mitogenomes.zip
- Nanoporeのシーケンスデータ ・・・メタゲノムとeDNAのデータが入っています。
https://suikou.fs.a.u-tokyo.ac.jp/yosh_data/2024jissyu/2024nanopore.zip
zipファイルを解凍し、中身の
X_XXX_1st.fq,X_XXX_2nd.fqファイルを「2024jissyu」フォルダにコピーしておく。_1stは三年生の皆さんがPCRしてナノポアシーケンスしたデータ、_2ndはデータが少なかったサンプルを研究室のスタッフで追加実験したデータ。・ eDNA解析: X-eDNA.fqのファイルを使用
・ メタゲノム解析(水・食品): X-food.fq, X-metagenome.fqのファイルを使用
B. クオリティチェック
FastQCは主にIllumina用のクオリティチェックツールなので、Nanoporeのデータに対しては適切な評価ができておらず、評価値の〇×は気にしなくてよいです。
FASTQCを実行するには…
- Windows
解凍したFastQCのフォルダの中の
run_fastqc.batを実行する。 - Mac
dmgファイルを開いて、FASTQCを実行すると、下記のエラーが出ると思う。
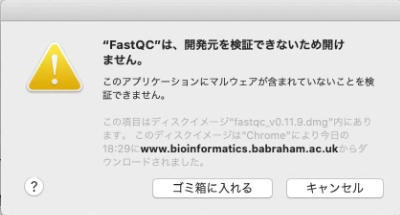
一度「キャンセル」して、「システム環境設定」を開き、 「プライバシーとセキュリティ」を開き、「このまま開く」をクリックしてFastQCを開く。



FastQCでシーケンスファイルを開く

Macでは下記のようにアクセスを許可する必要があるので、許可をする。

Ctrlキーを押しながらクリックすれば、複数のファイルを一度に選択できるので、必要なファイルを選択して開く。(ただし一度に複数選択するとメモリーエラーで落ちたりするので、その場合は一つずつ開くこと。)

結果のタブを開いてみると、次のようなQC結果が見られる。

Nanoporeのデータだとクオリティスコア10強(精度90%強)となるはずですが、リード全体のクオリティは比較的正しく計算できるみたいだけど、塩基ごとのクオリティはあまり正確ではなく、実際の塩基精度とは乖離があるようです。(ナノポアでQ20と出ていても、実際はQ12.5程度。https://labs.epi2me.io/quality-scores/)
リード長の分布が想定される長さになっているかなども確認すること。 (バクテリア16S全長は1.5 kbp程度、魚類ミトコンドリア12S MiFishは200 bp強)
C. BLAST
1. FASTQ→FASTA変換
FASTQファイルをメモ帳などで開いてみると次のように表示されます。

FASTQ形式は 塩基配列とクオリティが両方含まれるファイル形式になりますが、BLASTのプログラムはFASTQ形式では入力ファイルとして受け付けてくれません。BLASTが対応しているのは、塩基配列だけの情報であるFASTA形式になります。
FASTA形式は1つのシーケンスにつき、「>」 で始まる1行のヘッダ行と、2行目以降のACGTのシーケンス文字列で構成されます。

そこで、まずFASTQのクオリティを削除して、FASTA形式に変換します。
./seqkit fq2fa -o 1-metagehome_1st.fasta 1-metagehome_1st.fq
1-metagehome_1st.fq, 1-metagehome_1st.fastaは適当に変更すること。
もしリード数が多い場合は、後のblast検索で時間がかかるので、例えば500リードなどにダウンサンプリングしておく。(特にfood, metanogemサンプルはリード長が長く、blast検索に時間がかかるのでリード数を減らしておくことを推奨)
#例 ./seqkit sample -n 500 -o 1-metagehome_1st_500.fasta 1-metagehome_1st.fasta
コマンド入力時の便利キー
| カーソルの上・下キー | 前に入力したコマンドを呼び出す。 |
| (Win) Ctrl+Shift+V or 右クリック, (Mac) Cmd+V | 貼り付け |
| tabキー | ファイル名・コマンドの自動補完 |
| Ctrl+C | コマンド強制終了(blastを実行中に止めたい場合など) |
2. BLASTデータベース作成
makeblastdbコマンドによってBLASTのデータベースを作成します。ターミナルで下記のように入力します。
#Blastデータベースを作成 ./ncbi-blast-2.16.0+/bin/makeblastdb -in silva-SSU-LSU_PR2_NCBI-16S-mito-plastid_2023-04-18_rename_mitofish-2023-11-07.fasta -dbtype nucl
-in には入力となるFASTAファイルを指定します。
-dbtype はFASTAファイルがDNA配列であれば「nucl」、アミノ酸配列であれば「prot」を指定します。
3. BLAST検索
FASTA形式に変換したシーケンスデータをクエリーとして、バクテリア16SデータベースもしくはMitoFishデータベースで相同性検索を行います。ターミナルで次のように入力してください。
./ncbi-blast-2.16.0+/bin/blastn -db silva-SSU-LSU_PR2_NCBI-16S-mito-plastid_2023-04-18_rename_mitofish-2023-11-07.fasta -query 1-metagehome_1st_500.fasta -num_threads 16 -out 1-metagehome_1st_500.fasta.blastn
-db には、BLASTデータベースファイルのprefixを指定します。今回の場合は元となったSILVAもしくはMitoFishのFASTAファイルを指定すれば良いです。
-query にはデータベースに対して相同性検索を行いたい問い合わせ配列が含まれるFASTAファイルを指定します。ここではFASTA形式に変換したシーケンスデータを指定します。(ファイル名は適宜変更すること。)
-num_threads には並列計算時に使用するCPUの数を指定します。自分のPCで計算するときは、各自のPCに合わせて設定すること。
-out には出力ファイルの名前を書く。(拡張子はMEGANに読み込ませるために「.blastn」とし、ファイル名は出来れば「入力FASTAファイル名」+「.blastn」としておくとMEGANがFASTAファイルを認識してくれて情報がリッチになる。)
4. BLAST結果例
下記のように入力すると、結果ファイルの中身を先頭30行だけ見ることが出来ます。
## Windows (PowerShell)の場合 Get-Content -head 30 1-metagehome_1st_500.fasta.blastn ## Mac/Linuxの場合 head -n 30 1-metagehome_1st_500.fasta.blastn # 1-metagehome_1st_500.fasta.blastn は適当なファイル名に変更します。


D. MEGANによるBLAST結果集計
1.MEGANではLowest Common Ancestor (LCA)法によってtaxon (分類群)を推測している。LCA法とは、1つのリードがデータベース中の複数の配列にヒットした場合、ヒットした配列に共通の分類群を求める方法です。 http://bio4j.com/blog/2012/02/finding-the-lowest-common-ancestor-of-a-set-of-ncbi-taxonomy-nodes-with-bio4j/ ただ、MEGANのデフォルト値はIlluminaシーケンサー用(精度99.9%)に設定されているので、ナノポアのように80~90%程度の精度では変更する必要があります。

2.インストールしたMEGANを起動します。

3.MEGANが起動したら、「File」→「Import From BLAST」からBLAST結果を開きます。

4.ダイアログ右上のボタンをクリックして、BLASTの結果ファイルを選択し、「Open」をクリックします。(複数ファイルを選択できますが、結果がマージされてしまうので、ファイルを一つだけ選択します。}

5.「LCA Params」タブを開いて、Top Percent: の値を0.5に変更しておきます。このパラメータは、BLASTの結果の中で最もスコアの高いトップヒットからどの程度離れたヒットまで使用するかの閾値になります。ナノポアではシーケンス精度が悪く、無関係な生物も似たようなスコアでヒットしてしまうため、ほぼトップヒットしか使わないように厳しめに閾値を設定しておきます。それから、Min Score: の値をバクテリア16Sではリード長が1500bp程度なので1000、魚類12Sではリード長が200bp程度なので100などと指定し、スコアの低いリードをトリミングします。「Apply」を押すとファイルを読み込みます。

6.結果が表示されたら、 種名まで表示するために、「Rank」→「Species」を選択します。

↓

7.マウスホイールで拡大・縮小できます。もしくはメニューバーの下記のボタンをクリックしても拡大できます。

8.各生物種に割り当てられたBLASTの生データを見たい場合、ノードを右クリックし、「Inspector」を開けば見れます。本当にその生物はいるのか確認したくなった時などに、アライメント長や塩基配列そのものを確認するのに便利です。


9.BLASTファイルを読み込むときに設定したLCAのパラメータを後から変更したい場合は、「Options」→「Change LCA Parameters」から可能です。

10.MEGANで複数サンプルを比較する場合は、blastnファイルを複数回開いたら、開いたウィンドウをそのままにした状態で、「File」→「Compare」とクリックして比較したいサンプルを選択し、「Apply」をクリックします。

11.複数結果の表示を変更する場合、円グラフのボタンなどを押すと良いです。

12.ExcelやR等で解析する場合は、データをタブ区切りテキスト形式でExportする必要があります。Exportしたいノードをクリックして(全部Exportする場合は全部選択(WidnowsならCtrl+a)して)、「File」→「Export」→「CSV Format」をクリックします。

13.Exportするデータを「taxonPath_to_count」に変更します。(ほかのデータ形式でも勿論可)

14.適当な名前を付けて保存します。

E. データの転送、Excelでの解析
ExportしたファイルをExcelで開くには、Excelを起動しておき、ExportしたファイルをExcel上にドラッグアンドドロップすれば良いです。

Excelでデータの概要を把握するのに役立つテクニックとして、条件付き書式を設定することで、データの大小を一目でわかるようにできたりします。

そのほか、「データ」→「フィルター」を使ってみたり、グラフを描いてみたりするのが通常の解析の流れになるかと思います。
F. 最終日のプレゼンテーションの内容
各班次の内容について「目的」、「方法」、「結果」、「考察」の4つのパートを明確に区別してプレゼンテーションを作成してください。班ごとに発表し、発表時間は質疑応答を入れて30分です。
X班.食品の品種判別 by サンガー X班.三四郎池のeDNA X班.発酵食品のメタゲノム X班.三四郎池のメタゲノム
各テーマごとに例えば下記のような項目について考察をすること。インターネットを積極的に使用して調べることを推奨します。また、ある程度調べてもわからないことがあればTA・スタッフに聞いてみてください。
・X班.食品の品種判別 by サンガー
NCBIのデータベースとMitoFishのデータベースを比べて、ヒットした種が同じかどうか調べ、どちらのデータベースのほうが良さそうか考えてみる。
ネガティブコントロールでもPCR増幅してしまった理由を考えて、どうすればネガティブコントロールで増幅しないようにできるか、どうやって検証するか考える。
手法で詳しく説明して欲しい箇所:「DNA抽出」(使用したキットはDNeasy Blood & Tissue Kitsです。)
・X班.三四郎池のeDNA
検出された魚は三四郎池に棲息していそうな魚かどうか。
三年前の三四郎池のデータとも比較してみてください。http://suikou.fs.a.u-tokyo.ac.jp/yosh_data/2021jissyu/2021nanopore.zip (group2-12S-Sanshiroike1.fq)
手法で詳しく説明して欲しい箇所:「電気泳動、DNA精製」(使用したキットはFastGene™ Gel/PCRExtractionキットです。)
・X班.発酵食品のメタゲノム
今回発酵食品で検出されるバクテリアはほぼ1種類だと思うので、精度の悪いナノポアのリードの精度を向上させる方法を実践してみてください。具体的にはGeneiousでマルチプルアライメントを作成して、コンセンサス配列を作ることで、NCBIのBlastで一致率99%程度のヒットが得られるようになることを確認し、ナノポアのリードはどのような間違いが多いのか考察してみてください。
加工食品から検出された魚は妥当でしょうか。No Hitのリードを抜き出して、NCBIのBLASTにかけてみると何がヒットしますか?(リードを抜き出す例: ./seqkit grep -rp "1e2f600e-220b-4144-93b0-75ccd2c537de:35:1568:-1:bac-27F-BC05:bac-1492R" ./2022nanopore/y2022-group1-bacteria-16S-Sanshiro.fq)
手法で詳しく説明して欲しい箇所:「PCR」(使用したDNAポリメラーゼはrepliQa HiFi ToughMixです。AmpliTaq GoldやEx Taqといった他の酵素と比較して、どういった特徴があるでしょうか。)
・X班.三四郎池のメタゲノム
検出されたバクテリアは淡水環境で良く検出されているでしょうか?
二年前の三四郎池のデータとも比較してみてください。http://suikou.fs.a.u-tokyo.ac.jp/yosh_data/2021jissyu/2021nanopore.zip (group2-16S-Sanshiroike1.fq)
手法で詳しく説明して欲しい箇所:「ナノポアシーケンシング」(使用したライブラリー調整キットはSQK-LSK110です。公式マニュアル:http://suikou.fs.a.u-tokyo.ac.jp/yosh_data/2022jissyu/amplicons-by-ligation-sqk-lsk110-ACDE_9110_v110_revT_10Nov2020-minion.pdf )
G. 課題
来週のプレゼン資料の完成版をファイルに保存して、発表時に提出すること。