WindowsやMacから遺伝研のスパコンにお手軽にNGS解析ジョブを投げるツールとしてPortablePipelineを開発しました。当研究室で使用頻度の高い解析パイプラインが実行できます。解析サーバとしてはpython3とdockerもしくはsingularityがインストールされていればスパコンでなくても実行できます。実行手順を下記に紹介します。
まず、PortablePipelineのWebページを開いて、最新版をダウンロードしてください。
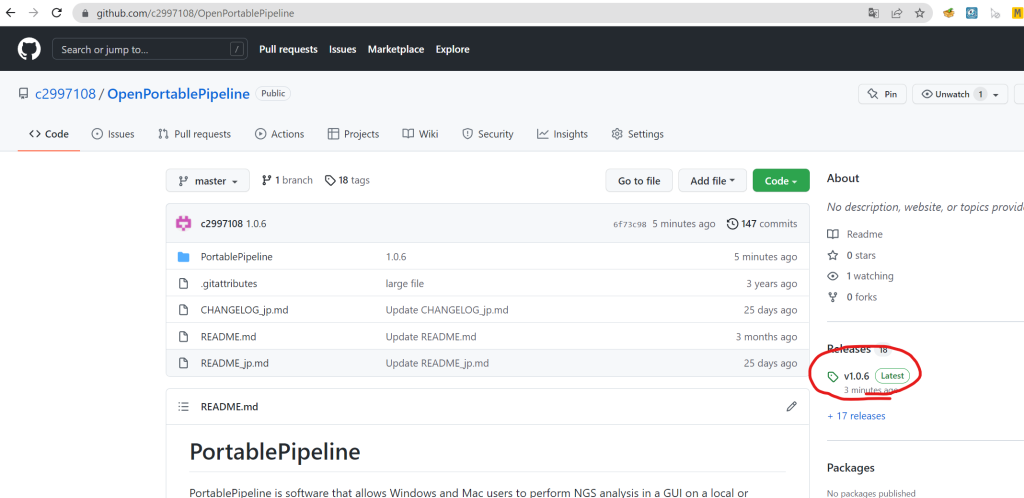
Windows、Macそれぞれで適切なファイルをダウンロードしてください。
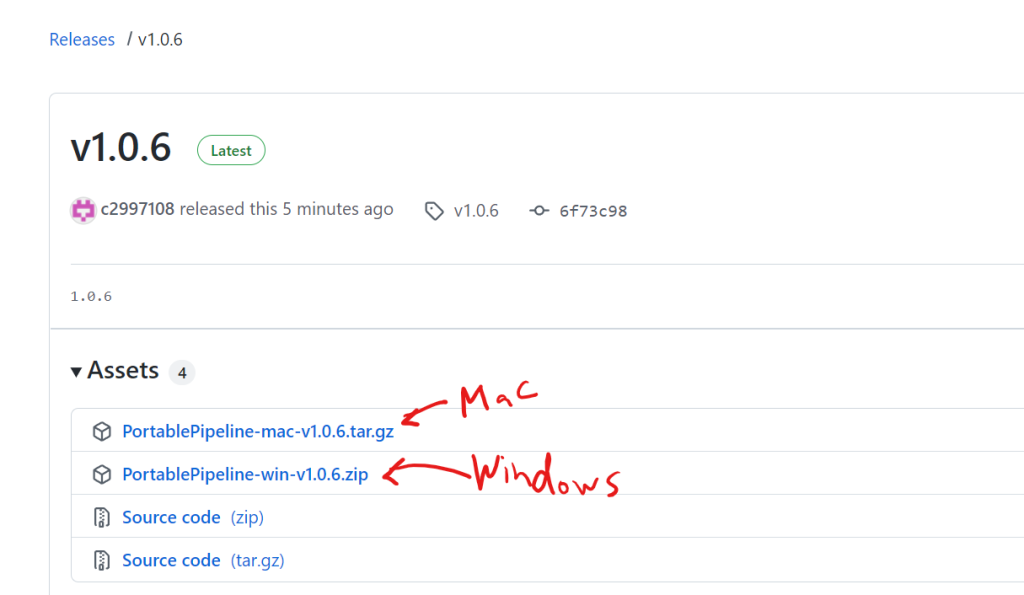
以下はWindowsでの操作方法を紹介します。まずはダウンロードしたzipファイルをすべて展開してください。
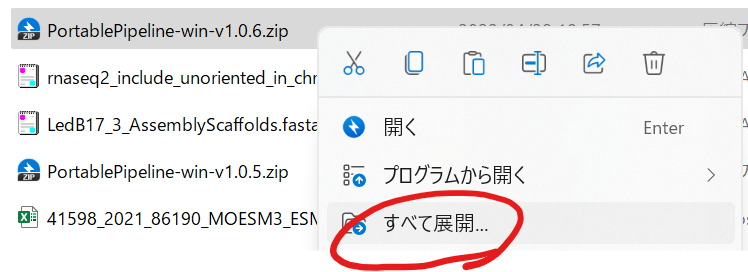
解凍して出来た「PortablePipeline.bat」というファイルを実行してください。
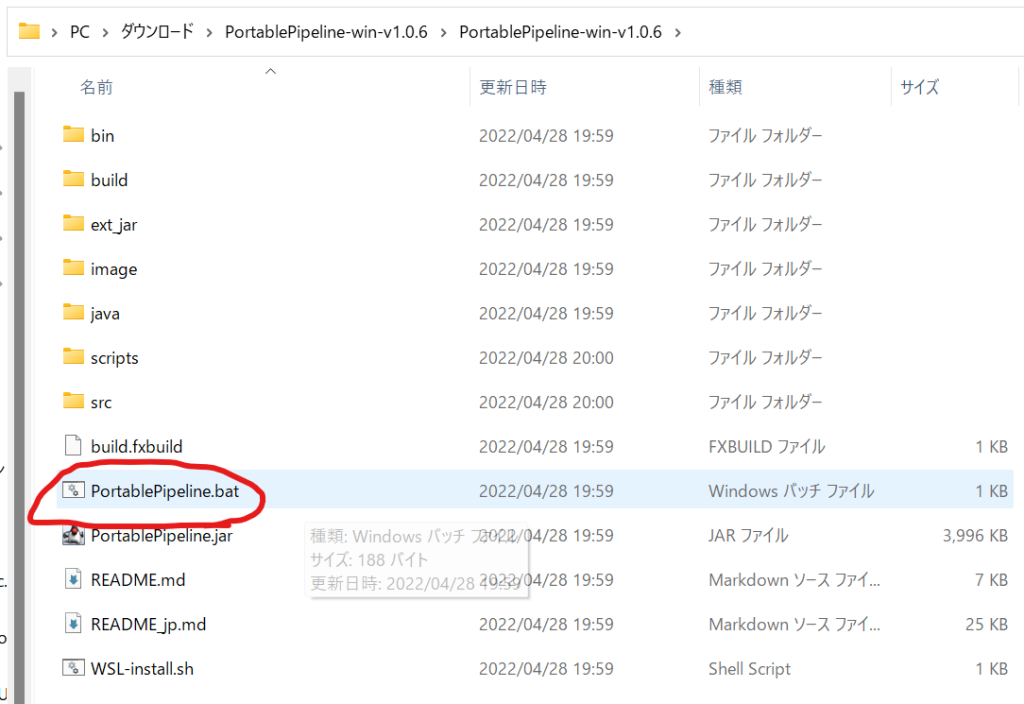
初回実行時にはMicrosoft Defenderが警告を表示しますが、怪しいソフトではないので(怪しい?)、「詳細情報」をクリックして
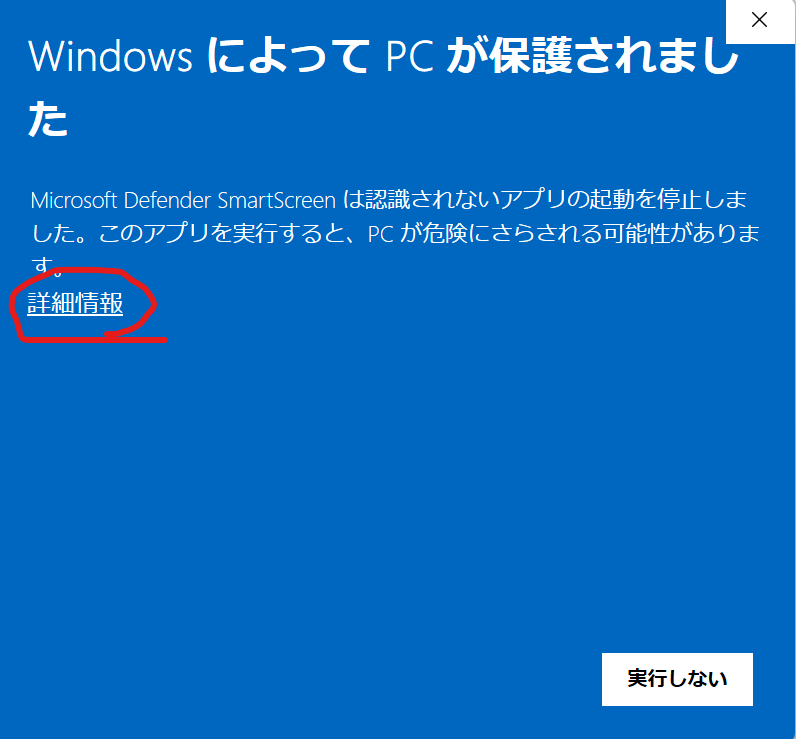
「実行」をクリックしてください。
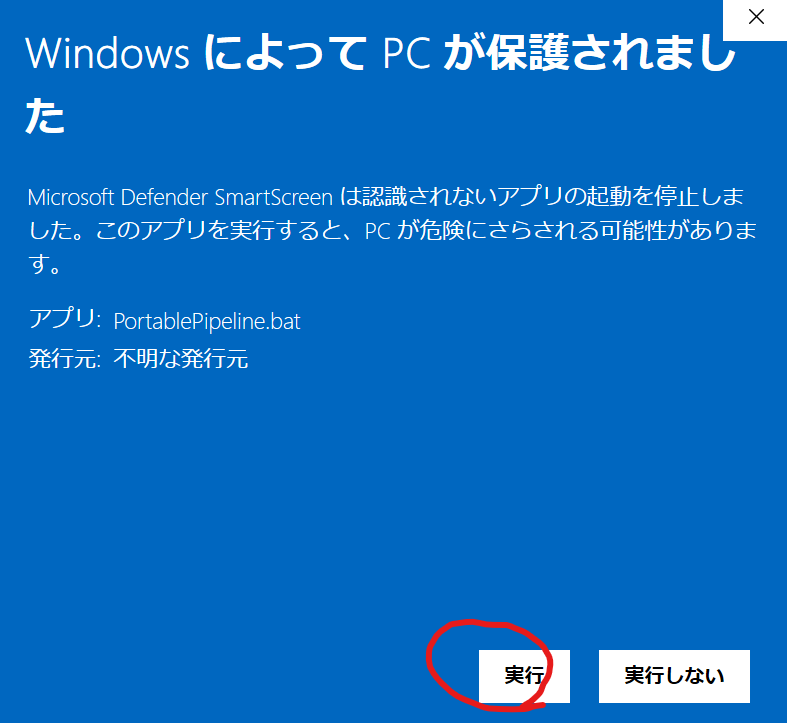
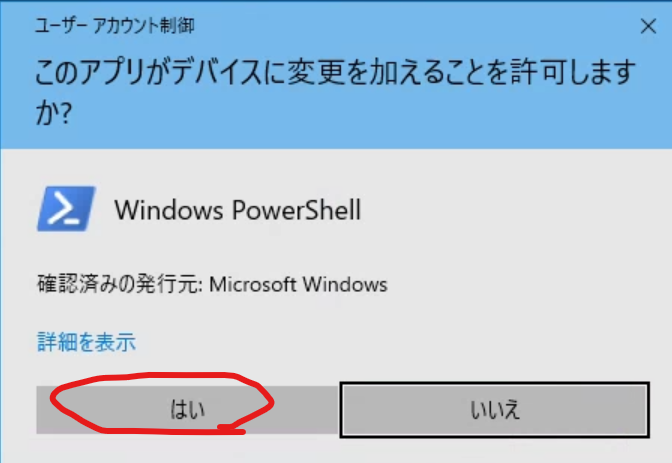
次に下記の確認画面が出てくるので、「はい」を選択します。本ツールはWindows環境下だと管理者権限で動かさないとシンボリックリンクファイル(ジャンクション)を作れないので管理者権限で動かすことに対する確認です。
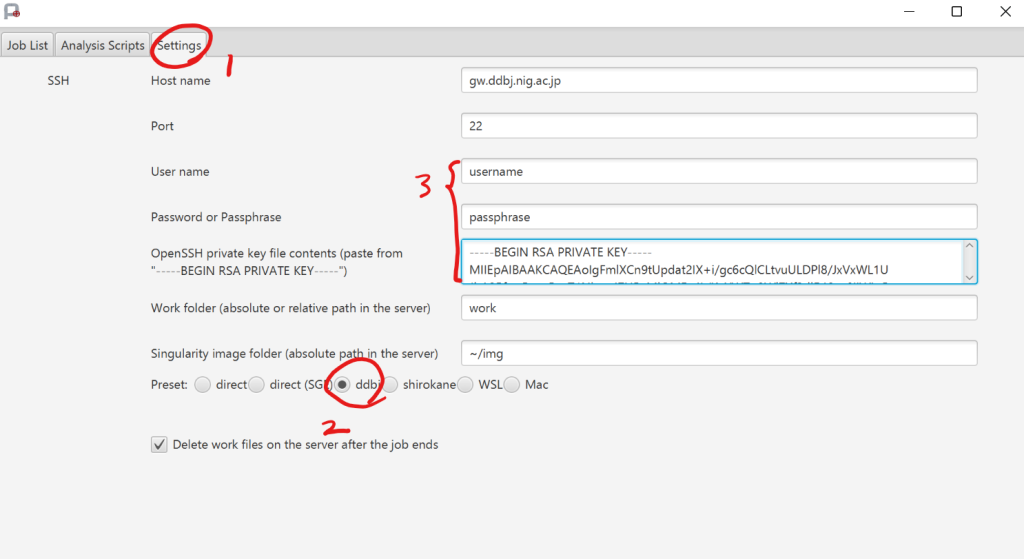
そうするとPortablePipelineが起動します。起動したらDDBJのスパコンに接続するためのアカウント情報を設定します。まだDDBJのアカウントを持っていない方はこちらから。「Settings」タブを開いて、「ddbj」を選択し、「User name」と「OpenSSH private key」と、もしOpenSSH private keyにパスフレーズを設定しているなら「Passphrase」にそれぞれ必要な情報を入力します。
今回はゲノムvsゲノムのドットプロットを描画するプログラムを実行してみます。ゲノムを比較する際にまずはゲノム全体のドットプロットを描いてみることが多く、個人的に使用頻度の高いツールです。
「Analysis Scripts」タブを開き、「post-assemble~dotplot-by-minimap2」を選択します。左側のアイコンをクリックすると説明ページに飛びます。(説明ページが十分でなくてすみません。パイプラインの中身のスクリプトはダウンロードしたscriptsフォルダに入っているので、詳しい方はソースコードをご覧ください。。。)このパイプラインは入力として2つのFASTAファイルを選択し、その2つのゲノムを比較したドットプロットを作成します。「input1」と「input2」をクリックして、それぞれFASTAファイルを選択してください。選択したら「Run」クリックしてください。CPU threadsとmemory limitはスパコンで実行する場合、とても重要なパラメーターです。大きくすれば解析プログラムがメモリ不足でこける可能性が減りますが、DDBJスパコンの稼働状況ページを見ると、ほぼいつもリソースが空いていない状況だと思うので、大きすぎる値を設定するといつまでたってもジョブが実行されません。また、基本的にはDDBJの epyc.q というキューで処理されるはずですが、この epyc.q のノードのメモリーは最大512GBのようなので、それ以上を指定すると永遠に実行されません。
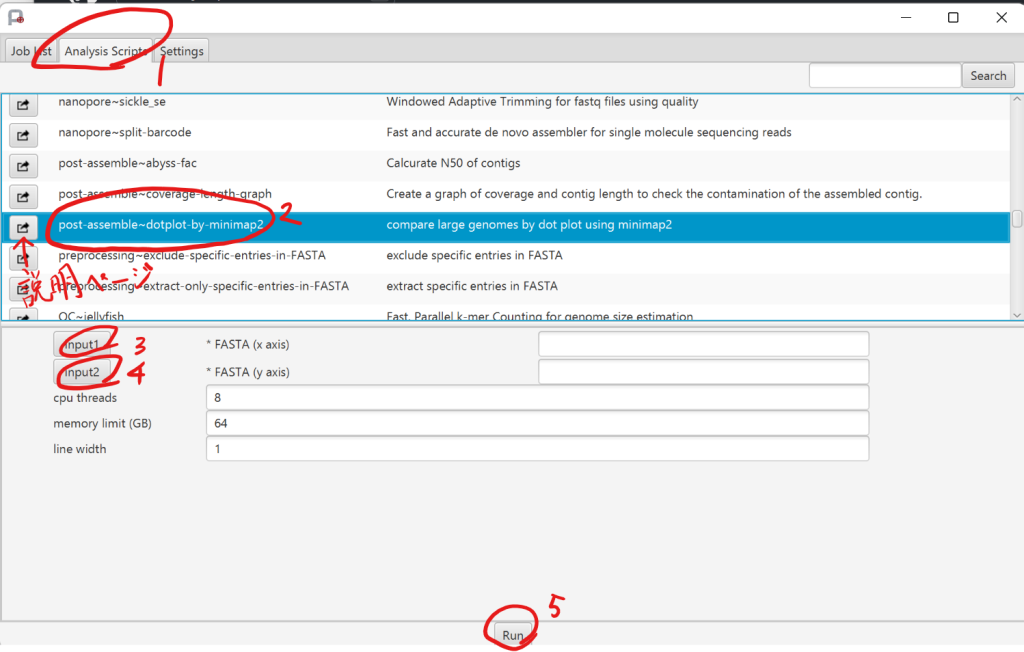
正しく情報が入力されていると、下記のように「Job List」が表示されます。「Auth fail」などと表示されると、スパコンのアカウントが正しく入力されていません。「running」のステータスになれば、PortablePipelineを閉じても大丈夫です。次にPortablePipelineを開いた時にジョブが終わっていればサーバにファイルを取りに行きます。そのまま開いていても大丈夫です。
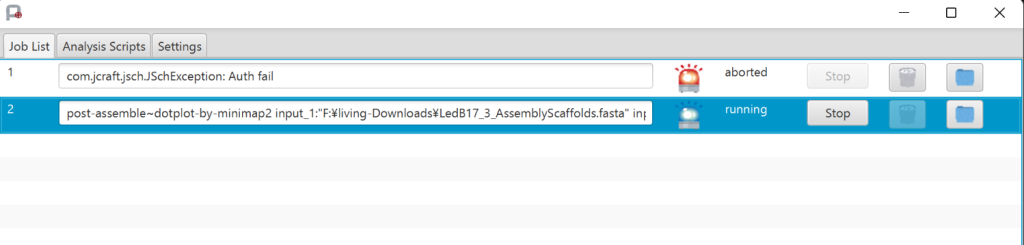
解析が終わるとステータスが「finished」になります。右端のフォルダーアイコンをクリックすると、解析結果を見ることが出来ます。
解析結果として下記のようなドットプロットが得られます。

他にもde novo RNA-seq解析用の「RNA-seq~Trinity-kallisto-sleuth」や、メタゲノム解析用の「metagenome~silva_SSU+LSU」などはFASTQファイルをセットするだけで最後まで解析してくれるので、そうした解析をこれから始めようという方には特におすすめです。